| Startseite |
Input
!
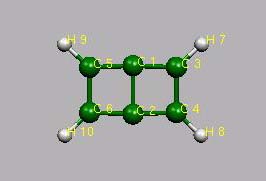
! Bicyclobuten
!
$CONTRL SCFTYP=RHF MULT=1 RUNTYP=OPTIMIZE COORD=ZMT $END
$SYSTEM TIMLIM=100 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Bicyclobuten
CNH 2
C1
C2 1 1.55
C3 1 1.38 2 87.8
C4 2 1.38 1 87.8 3 0.0 0
C5 1 1.38 2 87.8 4 -180.0 0
C6 2 1.38 1 87.8 3 180.0 0
H7 3 1.08 1 135.1 2 -180.0 0
H8 4 1.08 2 135.1 1 -180.0 0
H9 5 1.08 1 135.1 2 180.0 0
H10 6 1.08 2 135.1 1 180.0 0
$END
Output
TOTAL NUMBER OF SHELLS = 16
TOTAL NUMBER OF BASIS FUNCTIONS = 34
NUMBER OF ELECTRONS = 40
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 20
NUMBER OF OCCUPIED ORBITALS (BETA ) = 20
TOTAL NUMBER OF ATOMS = 10
THE NUCLEAR REPULSION ENERGY IS 191.6472791365
NSERCH= 6 ENERGY= -228.6797377
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C1 6.0 0.0000000 0.0000000 0.0000000
2 C1 6.0 0.0000000 0.0000000 0.0000000
3 C3 6.0 0.0000001 0.0000000 0.0000007
4 C3 6.0 -0.0000001 0.0000000 -0.0000007
5 C3 6.0 0.0000001 0.0000000 -0.0000007
6 C3 6.0 -0.0000001 0.0000000 0.0000007
7 H7 1.0 -0.0000028 0.0000000 0.0000046
8 H7 1.0 0.0000028 0.0000000 -0.0000046
9 H7 1.0 -0.0000028 0.0000000 -0.0000046
10 H7 1.0 0.0000028 0.0000000 0.0000046
MAXIMUM GRADIENT = 0.0000046 RMS GRADIENT = 0.0000020
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Bicyclobuten
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -0.7739718015
C3 6.0 1.3790625938 0.0000000000 -0.7221629769
H7 1.0 2.1742680250 0.0000000000 -1.4559201054
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 0.7739718015
C1 6.0 0.0000000000 0.0000000000 -0.7739718015
C3 6.0 -1.3790625938 0.0000000000 -0.7221629769
C3 6.0 1.3790625938 0.0000000000 0.7221629769
C3 6.0 -1.3790625938 0.0000000000 0.7221629769
C3 6.0 1.3790625938 0.0000000000 -0.7221629769
H7 1.0 -2.1742680250 0.0000000000 -1.4559201054
H7 1.0 2.1742680250 0.0000000000 1.4559201054
H7 1.0 -2.1742680250 0.0000000000 1.4559201054
H7 1.0 2.1742680250 0.0000000000 -1.4559201054
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
C1
C1 1 1.5479436
C3 1 1.3800354 2 87.8485156
C3 2 1.3800354 1 87.8485156 3 0.0000000 0
C3 1 1.3800354 2 87.8485156 4 180.0000000 0
C3 2 1.3800354 1 87.8485156 3 180.0000000 0
H7 3 1.0820126 1 135.1499681 2 180.0000000 0
H7 4 1.0820126 2 135.1499681 1 180.0000000 0
H7 5 1.0820126 1 135.1499681 2 180.0000000 0
H7 6 1.0820126 2 135.1499681 1 180.0000000 0
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C1 C1 C3 C3
1 C1 0.0000000 1.5479436 * 2.0347562 * 1.3800354 *
2 C1 1.5479436 * 0.0000000 1.3800354 * 2.0347562 *
3 C3 2.0347562 * 1.3800354 * 0.0000000 3.1134116
4 C3 1.3800354 * 2.0347562 * 3.1134116 0.0000000
5 C3 1.3800354 * 2.0347562 * 1.4443260 * 2.7581252 *
6 C3 2.0347562 * 1.3800354 * 2.7581252 * 1.4443260 *
7 H7 3.1144597 2.2787047 * 1.0820126 * 4.1677577
8 H7 2.2787047 * 3.1144597 4.1677577 1.0820126 *
9 H7 2.2787047 * 3.1144597 2.3187060 * 3.6282996
10 H7 3.1144597 2.2787047 * 3.6282996 2.3187060 *
C3 C3 H7 H7
1 C1 1.3800354 * 2.0347562 * 3.1144597 2.2787047 *
2 C1 2.0347562 * 1.3800354 * 2.2787047 * 3.1144597
3 C3 1.4443260 * 2.7581252 * 1.0820126 * 4.1677577
4 C3 2.7581252 * 1.4443260 * 4.1677577 1.0820126 *
5 C3 0.0000000 3.1134116 2.3187060 * 3.6282996
6 C3 3.1134116 0.0000000 3.6282996 2.3187060 *
7 H7 2.3187060 * 3.6282996 0.0000000 5.2334099
8 H7 3.6282996 2.3187060 * 5.2334099 0.0000000
9 H7 1.0820126 * 4.1677577 2.9118402 * 4.3485361
10 H7 4.1677577 1.0820126 * 4.3485361 2.9118402 *
H7 H7
1 C1 2.2787047 * 3.1144597
2 C1 3.1144597 2.2787047 *
3 C3 2.3187060 * 3.6282996
4 C3 3.6282996 2.3187060 *
5 C3 1.0820126 * 4.1677577
6 C3 4.1677577 1.0820126 *
7 H7 2.9118402 * 4.3485361
8 H7 4.3485361 2.9118402 *
9 H7 0.0000000 5.2334099
10 H7 5.2334099 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 191.6280583930
ELECTRONIC ENERGY = -420.3077961016
TOTAL ENERGY = -228.6797377087
------------------
MOLECULAR ORBITALS
------------------
1 2
3 4
5
-11.1968 -11.1968 -11.1964 -11.1964
-11.1485
BU AG
AU BG
AG
1 C 1 S 0.000000 0.006847 -0.007646
0.000000 0.703912
2 C 1 S 0.000000 -0.004208 0.004528
0.000000 0.013486
3 C 1 X 0.003331 0.000000
0.000000 -0.003384 0.000000
4 C 1 Y 0.000000 0.000000
0.000000 0.000000 0.000000
5 C 1 Z 0.000000 -0.000046 0.000049
0.000000 -0.000004
6 C 2 S 0.000000 0.006847
0.007646 0.000000 0.703912
7 C 2 S 0.000000 -0.004208 -0.004528
0.000000 0.013486
8 C 2 X 0.003331 0.000000
0.000000 0.003384 0.000000
9 C 2 Y 0.000000 0.000000
0.000000 0.000000 0.000000
10 C 2 Z 0.000000 0.000046 0.000049
0.000000 0.000004
11 C 3 S 0.497782 0.497756 0.497532
0.497558 -0.004583
12 C 3 S 0.009402 0.009115 0.013209
0.013574 -0.002849
13 C 3 X 0.001086 0.000861 0.000754
0.001018 -0.001927
14 C 3 Y 0.000000 0.000000 0.000000
0.000000 0.000000
15 C 3 Z -0.000853 -0.000836 0.002031
0.002100 0.000040
16 C 4 S -0.497782 0.497756 -0.497532
0.497558 -0.004583
17 C 4 S -0.009402 0.009115 -0.013209
0.013574 -0.002849
18 C 4 X 0.001086 -0.000861 0.000754
-0.001018 0.001927
19 C 4 Y 0.000000 0.000000 0.000000
0.000000 0.000000
20 C 4 Z -0.000853 0.000836 0.002031
-0.002100 -0.000040
21 C 5 S 0.497782 0.497756 -0.497532
-0.497558 -0.004583
22 C 5 S 0.009402 0.009115 -0.013209
-0.013574 -0.002849
23 C 5 X 0.001086 0.000861 -0.000754
-0.001018 -0.001927
24 C 5 Y 0.000000 0.000000 0.000000
0.000000 0.000000
25 C 5 Z 0.000853 0.000836 0.002031
0.002100 -0.000040
26 C 6 S -0.497782 0.497756 0.497532
-0.497558 -0.004583
27 C 6 S -0.009402 0.009115 0.013209
-0.013574 -0.002849
28 C 6 X 0.001086 -0.000861 -0.000754
0.001018 0.001927
29 C 6 Y 0.000000 0.000000 0.000000
0.000000 0.000000
30 C 6 Z 0.000853 -0.000836 0.002031
-0.002100 0.000040
31 H 7 S -0.001980 -0.001987 -0.001935 -0.001918
-0.000080
32 H 8 S 0.001980 -0.001987 0.001935
-0.001918 -0.000080
33 H 9 S -0.001980 -0.001987 0.001935
0.001918 -0.000080
34 H 10 S 0.001980 -0.001987 -0.001935
0.001918 -0.000080
6 7 8 9 10
-11.1484 -1.1647 -1.0182 -0.8533 -0.7619
AU AG BU AU BG
1 C 1 S 0.703470 -0.123672 0.000000 -0.120375 0.000000
2 C 1 S 0.019155 0.294208 0.000000 0.368490 0.000000
3 C 1 X 0.000000 0.000000 -0.188633 0.000000 0.221028
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 C 1 Z -0.003981 -0.085584 0.000000 0.078376 0.000000
6 C 2 S -0.703470 -0.123672 0.000000 0.120375 0.000000
7 C 2 S -0.019155 0.294208 0.000000 -0.368490 0.000000
8 C 2 X 0.000000 0.000000 -0.188633 0.000000 -0.221028
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 C 2 Z -0.003981 0.085584 0.000000 0.078376 0.000000
11 C 3 S 0.005247 -0.085650 -0.115232 0.086792 -0.100577
12 C 3 S 0.003407 0.214291 0.306555 -0.275348 0.307789
13 C 3 X 0.002038 0.072501 0.035354 -0.075847 0.007186
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.000000
15 C 3 Z -0.000020 0.054738 0.083599 0.090871 -0.123774
16 C 4 S -0.005247 -0.085650 0.115232 -0.086792 -0.100577
17 C 4 S -0.003407 0.214291 -0.306555 0.275348 0.307789
18 C 4 X 0.002038 -0.072501 0.035354 -0.075847 -0.007186
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 0.000000
20 C 4 Z -0.000020 -0.054738 0.083599 0.090871 0.123774
21 C 5 S -0.005247 -0.085650 -0.115232 -0.086792 0.100577
22 C 5 S -0.003407 0.214291 0.306555 0.275348 -0.307789
23 C 5 X -0.002038 0.072501 0.035354 0.075847 -0.007186
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.000000
25 C 5 Z -0.000020 -0.054738 -0.083599 0.090871 -0.123774
26 C 6 S 0.005247 -0.085650 0.115232 0.086792 0.100577
27 C 6 S 0.003407 0.214291 -0.306555 -0.275348 -0.307789
28 C 6 X -0.002038 -0.072501 0.035354 0.075847 0.007186
29 C 6 Y 0.000000 0.000000 0.000000 0.000000 0.000000
30 C 6 Z -0.000020 0.054738 -0.083599 0.090871 0.123774
31 H 7 S 0.000016 0.030779 0.063412 -0.097803 0.170328
32 H 8 S -0.000016 0.030779 -0.063412 0.097803 0.170328
33 H 9 S -0.000016 0.030779 0.063412 0.097803 -0.170328
34 H 10 S 0.000016 0.030779 -0.063412 -0.097803 -0.170328
11 12 13 14 15
-0.7551 -0.6435 -0.5844 -0.5679 -0.4972
AG BU AU AG BU
1 C 1 S 0.075853 0.000000 -0.095539 0.061886 0.000000
2 C 1 S -0.236549 0.000000 0.317352 -0.237374 0.000000
3 C 1 X 0.000000 -0.193862 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.361101
5 C 1 Z 0.123151 0.000000 0.068114 -0.195720 0.000000
6 C 2 S 0.075853 0.000000 0.095539 0.061886 0.000000
7 C 2 S -0.236549 0.000000 -0.317352 -0.237374 0.000000
8 C 2 X 0.000000 -0.193862 0.000000 0.000000 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.361101
10 C 2 Z -0.123151 0.000000 0.068114 0.195720 0.000000
11 C 3 S -0.074007 0.021156 -0.031197 -0.012747 0.000000
12 C 3 S 0.254493 -0.094157 0.094080 0.036502 0.000000
13 C 3 X -0.182281 0.244492 -0.240626 -0.066128 0.000000
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.304876
15 C 3 Z 0.004760 0.115535 -0.094883 0.321025 0.000000
16 C 4 S -0.074007 -0.021156 0.031197 -0.012747 0.000000
17 C 4 S 0.254493 0.094157 -0.094080 0.036502 0.000000
18 C 4 X 0.182281 0.244492 -0.240626 0.066128 0.000000
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 0.304876
20 C 4 Z -0.004760 0.115535 -0.094883 -0.321025 0.000000
21 C 5 S -0.074007 0.021156 0.031197 -0.012747 0.000000
22 C 5 S 0.254493 -0.094157 -0.094080 0.036502 0.000000
23 C 5 X -0.182281 0.244492 0.240626 -0.066128 0.000000
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.304876
25 C 5 Z -0.004760 -0.115535 -0.094883 -0.321025 0.000000
26 C 6 S -0.074007 -0.021156 -0.031197 -0.012747 0.000000
27 C 6 S 0.254493 0.094157 0.094080 0.036502 0.000000
28 C 6 X 0.182281 0.244492 0.240626 0.066128 0.000000
29 C 6 Y 0.000000 0.000000 0.000000 0.000000 0.304876
30 C 6 Z 0.004760 -0.115535 -0.094883 0.321025 0.000000
31 H 7 S 0.169386 -0.218087 0.249608 -0.112590 0.000000
32 H 8 S 0.169386 0.218087 -0.249608 -0.112590 0.000000
33 H 9 S 0.169386 -0.218087 -0.249608 -0.112590 0.000000
34 H 10 S 0.169386 0.218087 0.249608 -0.112590 0.000000
16 17 18 19 20
-0.4901 -0.4762 -0.2974 -0.2899 -0.2220
BG BU AG AG BG
1 C 1 S 0.000000 0.000000 0.014397 0.000000 0.000000
2 C 1 S 0.000000 0.000000 -0.091441 0.000000 0.000000
3 C 1 X 0.351681 0.238027 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.518064
5 C 1 Z 0.000000 0.000000 0.562140 0.000000 0.000000
6 C 2 S 0.000000 0.000000 0.014397 0.000000 0.000000
7 C 2 S 0.000000 0.000000 -0.091441 0.000000 0.000000
8 C 2 X -0.351681 0.238027 0.000000 0.000000 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 -0.518064
10 C 2 Z 0.000000 0.000000 -0.562140 0.000000 0.000000
11 C 3 S -0.015581 -0.008613 -0.036733 0.000000 0.000000
12 C 3 S 0.049069 0.048054 0.183843 0.000000 0.000000
13 C 3 X 0.290933 -0.148036 0.125215 0.000000 0.000000
14 C 3 Y 0.000000 0.000000 0.000000 0.460420 -0.321553
15 C 3 Z 0.049419 0.349484 0.145076 0.000000 0.000000
16 C 4 S -0.015581 0.008613 -0.036733 0.000000 0.000000
17 C 4 S 0.049069 -0.048054 0.183843 0.000000 0.000000
18 C 4 X -0.290933 -0.148036 -0.125215 0.000000 0.000000
19 C 4 Y 0.000000 0.000000 0.000000 -0.460420 0.321553
20 C 4 Z -0.049419 0.349484 -0.145076 0.000000 0.000000
21 C 5 S 0.015581 -0.008613 -0.036733 0.000000 0.000000
22 C 5 S -0.049069 0.048054 0.183843 0.000000 0.000000
23 C 5 X -0.290933 -0.148036 0.125215 0.000000 0.000000
24 C 5 Y 0.000000 0.000000 0.000000 0.460420 0.321553
25 C 5 Z 0.049419 -0.349484 -0.145076 0.000000 0.000000
26 C 6 S 0.015581 0.008613 -0.036733 0.000000 0.000000
27 C 6 S -0.049069 -0.048054 0.183843 0.000000 0.000000
28 C 6 X 0.290933 -0.148036 -0.125215 0.000000 0.000000
29 C 6 Y 0.000000 0.000000 0.000000 -0.460420 -0.321553
30 C 6 Z -0.049419 -0.349484 0.145076 0.000000 0.000000
31 H 7 S -0.215026 -0.108219 -0.204249 0.000000 0.000000
32 H 8 S -0.215026 0.108219 -0.204249 0.000000 0.000000
33 H 9 S 0.215026 -0.108219 -0.204249 0.000000 0.000000
34 H 10 S 0.215026 0.108219 -0.204249 0.000000 0.000000
21 22 23 24 25
0.2142 0.2587 0.4602 0.5055 0.5237
AU BU AU BG AG
1 C 1 S 0.000000 0.000000 -0.005325 0.000000 -0.129583
2 C 1 S 0.000000 0.000000 0.078291 0.000000 0.880241
3 C 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.595189 0.000000 0.613618 0.000000
5 C 1 Z 0.000000 0.000000 0.742455 0.000000 0.209990
6 C 2 S 0.000000 0.000000 0.005325 0.000000 -0.129583
7 C 2 S 0.000000 0.000000 -0.078291 0.000000 0.880241
8 C 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 C 2 Y 0.000000 0.595189 0.000000 -0.613618 0.000000
10 C 2 Z 0.000000 0.000000 0.742455 0.000000 -0.209990
11 C 3 S 0.000000 0.000000 -0.084623 0.000000 0.065947
12 C 3 S 0.000000 0.000000 0.495300 0.000000 -0.405341
13 C 3 X 0.000000 0.000000 0.144619 0.000000 -0.444435
14 C 3 Y 0.559867 -0.372993 0.000000 0.479857 0.000000
15 C 3 Z 0.000000 0.000000 0.157677 0.000000 0.167686
16 C 4 S 0.000000 0.000000 0.084623 0.000000 0.065947
17 C 4 S 0.000000 0.000000 -0.495300 0.000000 -0.405341
18 C 4 X 0.000000 0.000000 0.144619 0.000000 0.444435
19 C 4 Y 0.559867 -0.372993 0.000000 -0.479857 0.000000
20 C 4 Z 0.000000 0.000000 0.157677 0.000000 -0.167686
21 C 5 S 0.000000 0.000000 0.084623 0.000000 0.065947
22 C 5 S 0.000000 0.000000 -0.495300 0.000000 -0.405341
23 C 5 X 0.000000 0.000000 -0.144619 0.000000 -0.444435
24 C 5 Y -0.559867 -0.372993 0.000000 -0.479857 0.000000
25 C 5 Z 0.000000 0.000000 0.157677 0.000000 -0.167686
26 C 6 S 0.000000 0.000000 -0.084623 0.000000 0.065947
27 C 6 S 0.000000 0.000000 0.495300 0.000000 -0.405341
28 C 6 X 0.000000 0.000000 -0.144619 0.000000 0.444435
29 C 6 Y -0.559867 -0.372993 0.000000 0.479857 0.000000
30 C 6 Z 0.000000 0.000000 0.157677 0.000000 0.167686
31 H 7 S 0.000000 0.000000 -0.102281 0.000000 0.027119
32 H 8 S 0.000000 0.000000 0.102281 0.000000 0.027119
33 H 9 S 0.000000 0.000000 0.102281 0.000000 0.027119
34 H 10 S 0.000000 0.000000 -0.102281 0.000000 0.027119
26 27 28 29 30
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -689.2053684489
TWO ELECTRON ENERGY = 268.8975723473
NUCLEAR REPULSION ENERGY = 191.6280583930
------------------
TOTAL ENERGY = -228.6797377087
ELECTRON-ELECTRON POTENTIAL ENERGY = 268.8975723473
NUCLEUS-ELECTRON POTENTIAL ENERGY = -918.3742132118
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 191.6280583930
------------------
TOTAL POTENTIAL ENERGY = -457.8485824715
TOTAL KINETIC ENERGY = 229.1688447628
VIRIAL RATIO (V/T) = 1.9978657350
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -151.4102253564
BARE H ENERGY= -689.2053684489
ELECTRONIC ENERGY = -420.3077969027
KINETIC ENERGY= 229.1688447628
N-N REPULSION= 191.6280583930
TOTAL ENERGY= -228.6797385097
SIGMA PART(1+2)= -380.7352843558
(K,V1,2)= 221.5376810801 -833.6162111770 231.3432457411
PI PART(1+2)= -39.5725125469
(K,V1,2)= 7.6311636827 -84.7580020348 37.5543258051
SIGMA SKELETON, ERROR= -189.1072259628 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000549 -0.000357 -0.000221 -0.000431 1.000761
2 -0.000549 -0.000357 -0.000221 -0.000431 1.000761
3 0.500421 0.500327 0.500239 0.500342 -0.000380
4 0.500421 0.500327 0.500239 0.500342 -0.000380
5 0.500421 0.500327 0.500239 0.500342 -0.000380
6 0.500421 0.500327 0.500239 0.500342 -0.000380
7 -0.000147 -0.000149 -0.000129 -0.000127 -0.000001
8 -0.000147 -0.000149 -0.000129 -0.000127 -0.000001
9 -0.000147 -0.000149 -0.000129 -0.000127 -0.000001
10 -0.000147 -0.000149 -0.000129 -0.000127 -0.000001
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 1.000580 0.456589 0.216140 0.388503 0.182268
2 1.000580 0.456589 0.216140 0.388503 0.182268
3 -0.000290 0.263221 0.364292 0.257286 0.288621
4 -0.000290 0.263221 0.364292 0.257286 0.288621
5 -0.000290 0.263221 0.364292 0.257286 0.288621
6 -0.000290 0.263221 0.364292 0.257286 0.288621
7 0.000000 0.008485 0.027638 0.048463 0.120245
8 0.000000 0.008485 0.027638 0.048463 0.120245
9 0.000000 0.008485 0.027638 0.048463 0.120245
10 0.000000 0.008485 0.027638 0.048463 0.120245
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.161745 0.116639 0.180948 0.243817 0.426031
2 0.161745 0.116639 0.180948 0.243817 0.426031
3 0.296828 0.278244 0.224235 0.339590 0.286985
4 0.296828 0.278244 0.224235 0.339590 0.286985
5 0.296828 0.278244 0.224235 0.339590 0.286985
6 0.296828 0.278244 0.224235 0.339590 0.286985
7 0.122299 0.163437 0.185291 0.038502 0.000000
8 0.122299 0.163437 0.185291 0.038502 0.000000
9 0.122299 0.163437 0.185291 0.038502 0.000000
10 0.122299 0.163437 0.185291 0.038502 0.000000
16 17 18 19 20
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.301838 0.139367 0.661221 0.000000 0.557507
2 0.301838 0.139367 0.661221 0.000000 0.557507
3 0.231080 0.401353 0.093272 0.500000 0.221246
4 0.231080 0.401353 0.093272 0.500000 0.221246
5 0.231080 0.401353 0.093272 0.500000 0.221246
6 0.231080 0.401353 0.093272 0.500000 0.221246
7 0.118001 0.028963 0.076118 0.000000 0.000000
8 0.118001 0.028963 0.076118 0.000000 0.000000
9 0.118001 0.028963 0.076118 0.000000 0.000000
10 0.118001 0.028963 0.076118 0.000000 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99592 1.98985
2 C 1 S 1.12082 1.03937
3 C 1 X 0.95527 1.00240
4 C 1 Y 0.98354 0.98491
5 C 1 Z 0.97685 1.00833
6 C 2 S 1.99592 1.98985
7 C 2 S 1.12082 1.03937
8 C 2 X 0.95527 1.00240
9 C 2 Y 0.98354 0.98491
10 C 2 Z 0.97685 1.00833
11 C 3 S 1.99587 1.98959
12 C 3 S 1.15519 1.04909
13 C 3 X 0.93919 0.98519
14 C 3 Y 1.00823 1.00755
15 C 3 Z 0.94843 0.98741
16 C 4 S 1.99587 1.98959
17 C 4 S 1.15519 1.04909
18 C 4 X 0.93919 0.98519
19 C 4 Y 1.00823 1.00755
20 C 4 Z 0.94843 0.98741
21 C 5 S 1.99587 1.98959
22 C 5 S 1.15519 1.04909
23 C 5 X 0.93919 0.98519
24 C 5 Y 1.00823 1.00755
25 C 5 Z 0.94843 0.98741
26 C 6 S 1.99587 1.98959
27 C 6 S 1.15519 1.04909
28 C 6 X 0.93919 0.98519
29 C 6 Y 1.00823 1.00755
30 C 6 Z 0.94843 0.98741
31 H 7 S 0.93689 0.96875
32 H 8 S 0.93689 0.96875
33 H 9 S 0.93689 0.96875
34 H 10 S 0.93689 0.96875
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 5.0256702
2 0.2952792 5.0256702
3 -0.1155500 0.4839770 4.8327102
4 0.4839770 -0.1155500 0.0033917 4.8327102
5 0.4839770 -0.1155500 0.4881042 -0.0063148 4.8327102
6 -0.1155500 0.4839770 -0.0063148 0.4881042 0.0033917
7 0.0050888 -0.0177914 0.3805740 -0.0002377 -0.0194987
8 -0.0177914 0.0050888 -0.0002377 0.3805740 -0.0002437
9 -0.0177914 0.0050888 -0.0194987 -0.0002437 0.3805740
10 0.0050888 -0.0177914 -0.0002437 -0.0194987 -0.0002377
6 7 8 9 10
6 4.8327102
7 -0.0002437 0.5889199
8 -0.0194987 0.0000112 0.5889199
9 -0.0002377 0.0000107 0.0000555 0.5889199
10 0.3805740 0.0000555 0.0000107 0.0000112 0.5889199
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 6.032398 -0.032398 6.024858 -0.024858
2 C1 6.032398 -0.032398 6.024858 -0.024858
3 C3 6.046912 -0.046912 6.018825 -0.018825
4 C3 6.046912 -0.046912 6.018825 -0.018825
5 C3 6.046912 -0.046912 6.018825 -0.018825
6 C3 6.046912 -0.046912 6.018825 -0.018825
7 H7 0.936889 0.063111 0.968746 0.031254
8 H7 0.936889 0.063111 0.968746 0.031254
9 H7 0.936889 0.063111 0.968746 0.031254
10 H7 0.936889 0.063111 0.968746 0.031254
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.548 0.957 1 4 1.380 1.477 1 5 1.380 1.477
2 3 1.380 1.477 2 6 1.380 1.477 3 4 3.113 0.194
3 5 1.444 1.299 3 7 1.082 0.963 4 6 1.444 1.299
4 8 1.082 0.963 5 6 3.113 0.194 5 9 1.082 0.963
6 10 1.082 0.963
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.981 3.981 0.000
2 C1 3.981 3.981 0.000
3 C3 3.963 3.963 0.000
4 C3 3.963 3.963 0.000
5 C3 3.963 3.963 0.000
6 C3 3.963 3.963 0.000
7 H7 0.996 0.996 0.000
8 H7 0.996 0.996 0.000
9 H7 0.996 0.996 0.000
10 H7 0.996 0.996 0.000
| Startseite |