| Startseite | s.a. Acetylen |
Ein Dummy-Atom ist ein Punkt im Raum zur besseren Beschreibung (Bezugspunkt) der Geometrie mit einer Z-Matrix. Es hat keinen Einfluss auf die Berechnung.
Ein Problem, bei dem die Einführung eines Dummy-Atoms hilft, ist die lineare Anordnung von drei Atomen, bei der der Torsionswinkel nicht mehr eindeutig definiert ist.
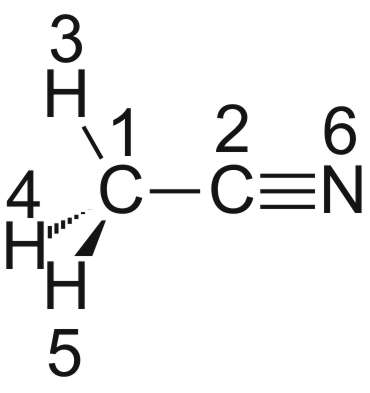
Beispiel: Acetonitril (Methylcyanid):
Definiert man die Z-Matrix wie folgt:
Acetonitril
CNV 3
C1
C2 1 R1
H3 1 R2 2 A1
H4 1 R2 2 A1 3 D1
H5 1 R2 2 A1 3 -D1
N6 2 R3 1 A2 3 D2
R1=1.5
R2=1.1
R3=1.3
A1=110.0
A2=180.0
D1=120.0
D2=0.0
erhält man im Output:
| --- ENCODED Z MATRIX --- COORD TYPE I J K L M N 1 1 2 1 2 1 5 1 3 2 5 1 2 4 1 4 1 5 2 4 1 2 6 3 4 1 2 5 7 1 3 1 8 2 3 1 2 9 3 3 1 2 5 10 1 6 2 11 2 6 2 1 12 3 6 2 1 5 ROUNDOFF ERROR IN BEND - STOP |
|
Umgeht man die Definition des Torsionswinkel entlang
der N-C-C-Bindung durch eine lange Bindungslänge
N6---C1
Acetonitril
CNV 3
C1
C2 1 R1
H3 1 R2 2 A1
H4 1 R2 2 A1 3 D1
H5 1 R2 2 A1 3 -D1
N6 1 R3 3 A2 2 D2
R1=1.5
R2=1.1
R3=2.7
A1=110.0
A2=110.0
D1=120.0
D2=0.0
wird die Rechnung erfolgreich beendet:
|
--- ENCODED Z MATRIX --- Alle Diederwinkel sind jetzt eindeutig |
|
THE DETERMINANT OF THE G MATRIX IS 10**( -8)
------------------------------------------
THE POINT GROUP IS CNV, NAXIS= 3, ORDER= 6
------------------------------------------
Eine Alternative dazu ist die Definition eines Dummy-Atoms:
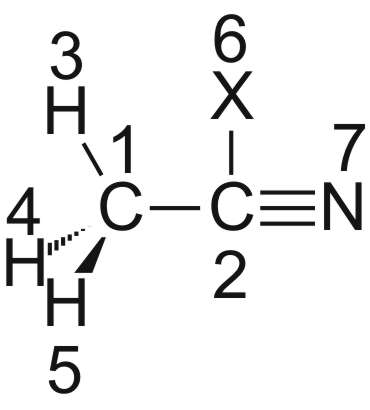
Acetonitril
CNV 3
C1
C2 1 R1
H3 1 R2 2 A1
H4 1 R2 2 A1 3 D1
H5 1 R2 2 A1 3 -D1
X6 2 R3 1 A2 3 D2
N7 2 R4 6 A3 1 D3
R1=1.5
R2=1.1
R3=1.0
R4=1.20
A1=110.0
A2=90.0
A3=90.0
D1=120.0
D2=0.0
D3=180.0
Den Diederwinkel D3 versteht man am besten, wenn man C1 nach oben an X6 schiebt:
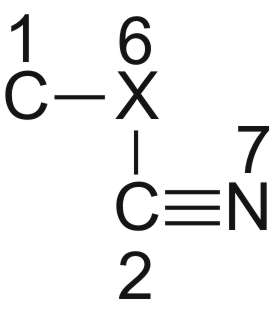
Man erhält dann:
YOUR FULLY SUBSTITUTED Z-MATRIX IS
C1
C2 1 1.5000000
H3 1 1.1000000 2 110.0000
H4 1 1.1000000 2 110.0000 3 120.0000 0
H5 1 1.1000000 2 110.0000 3 -120.0000 0
X6 2 1.0000000 1 90.0000 3 0.0000 0
N7 2 1.2000000 6 90.0000 1 180.0000 0
THE MOMENTS OF INERTIA ARE (AMU-ANGSTROM**2)
IXX= 58.273 IYY= 58.273 IZZ= 3.230
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
C1 6.0 0.0000000000 0.0000000000 -2.5181943539
C2 6.0 0.0000000000 0.0000000000 0.3163946277
H3 1.0 -0.9766688613 1.6916400899 -3.2291511423
H3 1.0 -0.9766688613 -1.6916400899 -3.2291511423
H3 1.0 1.9533377226 0.0000000000 -3.2291511423
N7 7.0 0.0000000000 0.0000000000 2.5840658130
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C1 C2 H3 H3
1 C1 0.0000000 1.5000000 * 1.1000000 * 1.1000000 *
2 C2 1.5000000 * 0.0000000 2.1421173 * 2.1421173 *
3 H3 1.1000000 * 2.1421173 * 0.0000000 1.7903549 *
4 H3 1.1000000 * 2.1421173 * 1.7903549 * 0.0000000
5 H3 1.1000000 * 2.1421173 * 1.7903549 * 1.7903549 *
6 N7 2.7000000 * 1.2000000 * 3.2452426 3.2452426
H3 N7
1 C1 1.1000000 * 2.7000000 *
2 C2 2.1421173 * 1.2000000 *
3 H3 1.7903549 * 3.2452426
4 H3 1.7903549 * 3.2452426
5 H3 0.0000000 3.2452426
6 N7 3.2452426 0.0000000
1NSERCH= 0
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -1.3325711612
C2 6.0 0.0000000000 0.0000000000 0.1674288388
H3 1.0 1.0336618829 0.0000000000 -1.7087933189
N7 7.0 0.0000000000 0.0000000000 1.3674288388
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -1.3325711612
C2 6.0 0.0000000000 0.0000000000 0.1674288388
H3 1.0 -0.5168309414 0.8951774495 -1.7087933189
H3 1.0 -0.5168309414 -0.8951774495 -1.7087933189
H3 1.0 1.0336618829 0.0000000000 -1.7087933189
N7 7.0 0.0000000000 0.0000000000 1.3674288388
NSERCH= 9 ENERGY= -131.5458580
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C1 6.0 0.0000000 0.0000000 -0.0000036
2 C2 6.0 0.0000000 0.0000000 -0.0000066
3 H3 1.0 -0.0000058 0.0000100 -0.0000017
4 H3 1.0 -0.0000058 -0.0000100 -0.0000017
5 H3 1.0 0.0000116 0.0000000 -0.0000017
6 N7 7.0 0.0000000 0.0000000 0.0000154
MAXIMUM GRADIENT = 0.0000154 RMS GRADIENT = 0.0000063
1 ***** EQUILIBRIUM GEOMETRY LOCATED
*****
Acetonitril
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -1.3228905720
C2 6.0 0.0000000000 0.0000000000 0.1616209928
H3 1.0 1.0189880462 0.0000000000 -1.6929065478
N7 7.0 0.0000000000 0.0000000000 1.3158957823
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C1 6.0 0.0000000000 0.0000000000 -1.3228905720
C2 6.0 0.0000000000 0.0000000000 0.1616209928
H3 1.0 -0.5094940231 0.8824695342 -1.6929065478
H3 1.0 -0.5094940231 -0.8824695342 -1.6929065478
H3 1.0 1.0189880462 0.0000000000 -1.6929065478
N7 7.0 0.0000000000 0.0000000000 1.3158957823
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C1 C2 H3 H3
1 C1 0.0000000 1.4845116 * 1.0840888 * 1.0840888 *
2 C2 1.4845116 * 0.0000000 2.1160362 * 2.1160362 *
3 H3 1.0840888 * 2.1160362 * 0.0000000 1.7649391 *
4 H3 1.0840888 * 2.1160362 * 1.7649391 * 0.0000000
5 H3 1.0840888 * 2.1160362 * 1.7649391 * 1.7649391 *
6 N7 2.6387864 * 1.1542748 * 3.1766693 3.1766693
H3 N7
1 C1 1.0840888 * 2.6387864 *
2 C2 2.1160362 * 1.1542748 *
3 H3 1.7649391 * 3.1766693
4 H3 1.7649391 * 3.1766693
5 H3 0.0000000 3.1766693
6 N7 3.1766693 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 58.1957607789
ELECTRONIC ENERGY = -189.7416187723
TOTAL ENERGY = -131.5458579934
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-15.5511 -11.2647 -11.2178 -1.1724 -0.9887
A1 A1 A1 A1 A1
1 C 1 S 0.000007 0.995120 -0.001795 -0.036746 -0.204763
2 C 1 S -0.000458 0.024353 -0.002817 0.118757 0.598561
3 C 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 C 1 Z -0.000300 0.000476 -0.002208 0.048201 0.014842
6 C 2 S 0.000299 0.002232 0.995645 -0.163852 -0.040868
7 C 2 S -0.007138 -0.003515 0.020000 0.369135 0.123956
8 C 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 C 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 C 2 Z -0.005522 0.003484 0.001956 0.181663 -0.226873
11 H 3 S 0.000060 -0.004479 -0.000230 0.015929 0.160432
12 H 4 S 0.000060 -0.004479 -0.000230 0.015929 0.160432
13 H 5 S 0.000060 -0.004479 -0.000230 0.015929 0.160432
14 N 6 S 0.996463 -0.000007 -0.000519 -0.187676 0.057726
15 N 6 S 0.018868 -0.000611 -0.004812 0.614088 -0.181464
16 N 6 X 0.000000 0.000000 0.000000 0.000000 0.000000
17 N 6 Y 0.000000 0.000000 0.000000 0.000000 0.000000
18 N 6 Z -0.006685 0.000387 0.001487 -0.221811 0.034103
6 7 8 9 10
-0.6444 -0.5962 -0.5962 -0.4713 -0.4110
A1 E E A1 E
1 C 1 S 0.033846 0.000000 0.000000 -0.011822 0.000000
2 C 1 S -0.119957 0.000000 0.000000 0.034604 0.000000
3 C 1 X 0.000000 0.000000 0.559911 0.000000 0.000000
4 C 1 Y 0.000000 0.559911 0.000000 0.000000 -0.176734
5 C 1 Z 0.478857 0.000000 0.000000 0.198487 0.000000
6 C 2 S -0.135261 0.000000 0.000000 0.060377 0.000000
7 C 2 S 0.479313 0.000000 0.000000 -0.170463 0.000000
8 C 2 X 0.000000 0.000000 0.201947 0.000000 0.000000
9 C 2 Y 0.000000 0.201947 0.000000 0.000000 0.573773
10 C 2 Z -0.309639 0.000000 0.000000 -0.342908 0.000000
11 H 3 S -0.174400 0.376366 -0.217295 -0.049871 -0.169735
12 H 4 S -0.174400 -0.376366 -0.217295 -0.049871 0.169735
13 H 5 S -0.174400 0.000000 0.434591 -0.049871 0.000000
14 N 6 S 0.076793 0.000000 0.000000 -0.116590 0.000000
15 N 6 S -0.311381 0.000000 0.000000 0.663623 0.000000
16 N 6 X 0.000000 0.000000 0.124286 0.000000 0.000000
17 N 6 Y 0.000000 0.124286 0.000000 0.000000 0.633004
18 N 6 Z -0.114111 0.000000 0.000000 0.665084 0.000000
11 12 13 14 15
-0.4110 0.3533 0.3533 0.5535 0.6846
E E E A1 E
1 C 1 S 0.000000 0.000000 0.000000 0.105958 0.000000
2 C 1 S 0.000000 0.000000 0.000000 -0.649509 0.000000
3 C 1 X -0.176734 0.000000 0.151319 0.000000 1.105958
4 C 1 Y 0.000000 0.151319 0.000000 0.000000 0.000000
5 C 1 Z 0.000000 0.000000 0.000000 -0.919423 0.000000
6 C 2 S 0.000000 0.000000 0.000000 -0.174883 0.000000
7 C 2 S 0.000000 0.000000 0.000000 1.247358 0.000000
8 C 2 X 0.573773 0.000000 0.808256 0.000000 -0.324962
9 C 2 Y 0.000000 0.808256 0.000000 0.000000 0.000000
10 C 2 Z 0.000000 0.000000 0.000000 -0.346629 0.000000
11 H 3 S 0.097997 -0.269717 0.155721 0.002348 0.480442
12 H 4 S 0.097997 0.269717 0.155721 0.002348 0.480442
13 H 5 S -0.195993 0.000000 -0.311443 0.002348 -0.960884
14 N 6 S 0.000000 0.000000 0.000000 0.055294 0.000000
15 N 6 S 0.000000 0.000000 0.000000 -0.371488 0.000000
16 N 6 X 0.633004 0.000000 -0.797817 0.000000 0.195413
17 N 6 Y 0.000000 -0.797817 0.000000 0.000000 0.000000
18 N 6 Z 0.000000 0.000000 0.000000 0.470841 0.000000
16 17 18
0.6846 0.6928 1.3106
E A1 A1
1 C 1 S 0.000000 -0.199524 -0.066193
2 C 1 S 0.000000 1.400367 0.563118
3 C 1 X 0.000000 0.000000 0.000000
4 C 1 Y 1.105958 0.000000 0.000000
5 C 1 Z 0.000000 -0.549942 0.418645
6 C 2 S 0.000000 0.016126 -0.058311
7 C 2 S 0.000000 -0.126989 0.796831
8 C 2 X 0.000000 0.000000 0.000000
9 C 2 Y -0.324962 0.000000 0.000000
10 C 2 Z 0.000000 -0.043172 1.538420
11 H 3 S -0.832150 -0.755179 -0.075946
12 H 4 S 0.832150 -0.755179 -0.075946
13 H 5 S 0.000000 -0.755179 -0.075946
14 N 6 S 0.000000 -0.010466 0.105302
15 N 6 S 0.000000 0.091728 -1.234802
16 N 6 X 0.000000 0.000000 0.000000
17 N 6 Y 0.195413 0.000000 0.000000
18 N 6 Z 0.000000 -0.096462 1.079056
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -292.8336941865
TWO ELECTRON ENERGY = 103.0920754142
NUCLEAR REPULSION ENERGY = 58.1957607789
------------------
TOTAL ENERGY = -131.5458579934
ELECTRON-ELECTRON POTENTIAL ENERGY = 103.0920754142
NUCLEUS-ELECTRON POTENTIAL ENERGY = -424.2667129768
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 58.1957607789
------------------
TOTAL POTENTIAL ENERGY = -262.9788767838
TOTAL KINETIC ENERGY = 131.4330187904
VIRIAL RATIO (V/T) = 2.0008585301
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.000002 2.002517 -0.000429 0.064531 1.082934
2 -0.001882 -0.000628 2.001349 0.725356 0.329508
3 0.000000 -0.000629 -0.000003 0.003092 0.171129
4 0.000000 -0.000629 -0.000003 0.003092 0.171129
5 0.000000 -0.000629 -0.000003 0.003092 0.171129
6 2.001884 -0.000002 -0.000911 1.200837 0.074170
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.811445 0.991489 0.991489 0.107918 0.069182
2 0.705781 0.153544 0.153544 0.232714 0.807634
3 0.099042 0.403829 0.134610 0.006248 0.059165
4 0.099042 0.403829 0.134610 0.006248 0.059165
5 0.099042 0.000000 0.538439 0.006248 0.000000
6 0.185648 0.047309 0.047309 1.640623 1.004855
11
2.000000
1 0.069182
2 0.807634
3 0.019722
4 0.019722
5 0.078886
6 1.004855
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99519 1.98853
2 C 1 S 1.16812 0.99315
3 C 1 X 1.06067 1.07487
4 C 1 Y 1.06067 1.07487
5 C 1 Z 0.90560 0.95479
6 C 2 S 1.99697 1.99160
7 C 2 S 1.06333 1.04361
8 C 2 X 0.96118 0.96333
9 C 2 Y 0.96118 0.96333
10 C 2 Z 0.93191 1.00591
11 H 3 S 0.89620 0.93418
12 H 4 S 0.89620 0.93418
13 H 5 S 0.89620 0.93418
14 N 6 S 1.99848 1.99797
15 N 6 S 1.76799 1.62639
16 N 6 X 1.05216 1.05250
17 N 6 Y 1.05216 1.05250
18 N 6 Z 1.33578 1.41412
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 4.6639776
2 0.3831005 4.8637764
3 0.3842659 -0.0222571 0.5723939
4 0.3842659 -0.0222571 -0.0189683 0.5723939
5 0.3842659 -0.0222571 -0.0189683 -0.0189683 0.5723939
6 -0.0096214 0.7344483 -0.0002618 -0.0002618 -0.0002618
6
6 6.4825375
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C1 6.190254 -0.190254 6.086208 -0.086208
2 C2 5.914554 0.085446 5.967784 0.032216
3 H3 0.896204 0.103796 0.934177 0.065823
4 H3 0.896204 0.103796 0.934177 0.065823
5 H3 0.896204 0.103796 0.934177 0.065823
6 N7 7.206579 -0.206579 7.143477 -0.143477
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.485 1.024 1 3
1.084 0.970 1 4 1.084 0.970
1 5 1.084 0.970 2 6
1.154 2.936
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C1 3.947 3.947 0.000
2 C2 3.968 3.968 0.000
3 H3 0.989 0.989 0.000
4 H3 0.989 0.989 0.000
5 H3 0.989 0.989 0.000
6 N7 2.995 2.995 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y
Z (BOHR) CHARGE
0.000000 0.000000 -0.028886
0.00 (A.U.)
DX DY
DZ /D/ (DEBYE)
0.000000 0.000000 -3.135650 3.135650
...... END OF PROPERTY EVALUATION ......
Die Geometrie-Optimierung erfolgt dann offensichtlich
in kartesischen Koordinaten. Die Koordinaten werden nicht als Z-Matrix
ausgegeben, d.h. man bekommt keine Winkelangaben. Das ist der Preis, den man für
die Einführung von Dummy-Atomen zahlen muss.
| Seitenanfang |