Grundzustand:
Erster angeregter Zustand:
0.98 eV = 22.5 kcal/mol
Zweiter angeregter Zustand:
762 nm = 13123 cm-1, 1.63 eV = 37.5 kcal/mol
Literaturangaben
Grundzustand:
![]() , Re = 1.21 A, De
= 5.21 eV, De = 4.839E-6,
, Re = 1.21 A, De
= 5.21 eV, De = 4.839E-6,
![]() = 1580 cm-1
= 1580 cm-1
Erster angeregter Zustand:
![]() :
Re = 1.2156 A, De = 4.86E-6, n00
= 7882.39 cm-1; 1270 nm = 7874 cm-1
:
Re = 1.2156 A, De = 4.86E-6, n00
= 7882.39 cm-1; 1270 nm = 7874 cm-1
0.98 eV = 22.5 kcal/mol
Zweiter angeregter Zustand:
![]() :
Re = 1.22688 A, De = 5.351E-6, n00
= 13120.91 cm-1,
:
Re = 1.22688 A, De = 5.351E-6, n00
= 13120.91 cm-1,
762 nm = 13123 cm-1, 1.63 eV = 37.5 kcal/mol
Ergebnisse
|
Total Energy /a.u. |
Bond Distance /Å |
|||||
| Triplet | Singlet | Difference | Triplet | Singlet | ||
| STO-6G | -149.05220 | -148.70862 | 0.34358 9.349 eV |
1.21757 | 1.271 | |
| STO-6G MP2 | -149.05219 | -149.16375 | 0.11156 3.036 eV |
1.31 | 1.351 | |
| STO-6G MCSCF | -149.17564 | -149.14276 | 0.03288 0.895 eV |
1.2928 | 1.317 | |
| DZV | -149.58947 | -149.24916 | 0.34031 9.261 eV |
1.207 | 1.284 | |
| DZV MP2 | -149.8278 | -149.79020 | 0.0376 1.023 eV |
1.36 | 1.318 | |
| DZV MCSCF 81) |
-149.735649 | -149.69147 | 0.044179 1.2022 eV |
1.3076 | 1.3230 | |
| DZV MCSCF 122) |
-149.777437 | |||||
| DZV MCSCF 14 |
-149.7288588 | 1.3273 | ||||
| TZV | -149.61026 | 1.209 | ||||
| TZV MP2 | -149.88182 | -149.8721 | 9.72E-3 0.2645 eV |
1.34 | 1.429 | |
| TZV 3df diff-sp | -149.68872 | -149.34788 | 0.34084 9.2749 eV |
1.158 | 1.217 | |
| cc-pVQZ MCSCF 6 |
-149.730182 | 1.224 | ||||
| cc-pVQZ MCSCF 8 |
-149.74128 | 1.225 | ||||
| cc-pVQZ MCSCF 5 |
-149.721171 | 1.18629 | ||||
1) Für Triplett: $DET
NCORE=4 NACT=10 NELS=8 NSTATE=1
$DRT NMCC=4 NDOC=3 NVAL=5 NALP=2
Für Singulett: $DET
NCORE=4 NACT=9 NELS=8 NSTATE=1
$DRT NMCC=4 NDOC=4 NVAL=5
2)Für Triplett: $DET NCORE=2 NACT=12 NELS=12 NSTATE=1
$DRT NMCC=2 NDOC=5 NVAL=5 NALP=2
STO-6G MULT=3 SCFTYP=UHF RUNTYP=OPTIMIZE
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
O
3 S 1 1355.584234 1.459053 ( 0.009164)
3 S 2 248.544886 2.202176 ( 0.049361)
3 S 3 69.533902 2.892385 ( 0.168538)
3 S 4 23.886772 2.853580 ( 0.370563)
3 S 5 9.275933 1.577736 ( 0.416492)
3 S 6 3.820341 0.253831 ( 0.130334)
4 L 7 52.187762 -0.183398 ( -0.013253) 0.751716 ( 0.003760)
4 L 8 10.329320 -0.192968 ( -0.046992) 0.994565 ( 0.037679)
4 L 9 3.210345 -0.057750 ( -0.033785) 1.065175 ( 0.173897)
4 L 10 1.235135 0.208956 ( 0.250242) 0.775885 (
0.418036)
4 L 11 0.536420 0.265853 ( 0.595117) 0.278668 (
0.425860)
4 L 12 0.245881 0.059902 ( 0.240706) 0.025102 (
0.101708)
L bedeutet s- und p-AO's. 4., 5. und 6.Spalte sind s-AO's, 7. und 8.Spalte sind die Koeffizienten für die p-AO's (Exponenten für beide gleich; in Klammern normierte Koeffizienten).
TOTAL NUMBER OF SHELLS = 4
TOTAL NUMBER OF BASIS FUNCTIONS = 10
NUMBER OF ELECTRONS = 16
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 3
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 9
NUMBER OF OCCUPIED ORBITALS (BETA ) = 7
TOTAL NUMBER OF ATOMS = 2
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
O1 O1
1 O1 0.0000000 1.2175702 *
2 O1 1.2175702 * 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 27.8155162052
ELECTRONIC ENERGY = -176.8677177263
TOTAL ENERGY = -149.0522015211
SPIN SZ =1.000
S-SQUARED =2.004
------------------
MOLECULAR ORBITALS
------------------
**** ALPHA SET ****
1 2
3 4
5
-20.7057
-20.7054 -1.6204 -1.1071 -0.7184
A2U A1G
A1G A2U EU
1 O 1 S 0.704551 0.705025 -0.162658 -0.178923 0.000000
2 O 1 S 0.012691 0.007943 0.583808 0.793231
0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000
0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.000000
0.660068
5 O 1 Z -0.004022 -0.000300 -0.154221 0.128671 0.000000
6 O 2 S -0.704551 0.705025 -0.162658 0.178923 0.000000
7 O 2 S -0.012691 0.007943 0.583808 -0.793231 0.000000
8 O 2 X 0.000000 0.000000 0.000000 0.000000
0.000000
9 O 2 Y 0.000000 0.000000 0.000000 0.000000
0.660068
10 O 2 Z -0.004022 0.000300 0.154221 0.128671 0.000000
6 7
8 9
10
-0.7184
-0.6126 -0.4261 -0.4261 0.6489
EU A1G
EG EG
A2U
1 O 1 S 0.000000 -0.056666 0.000000 0.000000
0.080984
2 O 1 S 0.000000 0.309939 0.000000 0.000000
-0.542070
3 O 1 X 0.660068 0.000000 0.765888 0.000000
0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.765888
0.000000
5 O 1 Z 0.000000 0.618084 0.000000 0.000000
0.943931
6 O 2 S 0.000000 -0.056666 0.000000 0.000000 -0.080984
7 O 2 S 0.000000 0.309939 0.000000 0.000000
0.542070
8 O 2 X 0.660068 0.000000 -0.765888 0.000000
0.000000
9 O 2 Y 0.000000 0.000000 0.000000 -0.765888
0.000000
10 O 2 Z 0.000000 -0.618084 0.000000 0.000000 0.943931
**** BETA SET ****
1
2 3
4 5
-20.6738 -20.6733
-1.4888 -0.9134 -0.5606
A1G
A2U A1G
A2U A1G
1 O 1 S 0.705328 0.704990 -0.158758 -0.172382 -0.063540
2 O 1 S 0.006783 0.010580 0.569032 0.759700
0.336323
3 O 1 X 0.000000 0.000000 0.000000 0.000000
0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.000000
0.000000
5 O 1 Z -0.000444 -0.003475 -0.182331 0.184433 0.610383
6 O 2 S 0.705328 -0.704990 -0.158758 0.172382 -0.063540
7 O 2 S 0.006783 -0.010580 0.569032 -0.759700 0.336323
8 O 2 X 0.000000 0.000000 0.000000 0.000000
0.000000
9 O 2 Y 0.000000 0.000000 0.000000 0.000000
0.000000
10 O 2 Z 0.000444 -0.003475 0.182331 0.184433 -0.610383
6 7
8 9
10
-0.4454
-0.4454 0.2573 0.2573 0.7378
EU EU
EG EG
A2U
1 O 1 S 0.000000 0.000000 0.000000 0.000000
0.090762
2 O 1 S 0.000000 0.000000 0.000000 0.000000 -0.588184
3 O 1 X 0.660068 0.000000 0.765888 0.000000
0.000000
4 O 1 Y 0.000000 0.660068 0.000000 0.765888
0.000000
5 O 1 Z 0.000000 0.000000 0.000000 0.000000
0.934639
6 O 2 S 0.000000 0.000000 0.000000 0.000000 -0.090762
7 O 2 S 0.000000 0.000000 0.000000 0.000000
0.588184
8 O 2 X 0.660068 0.000000 -0.765888 0.000000 0.000000
9 O 2 Y 0.000000 0.660068 0.000000 -0.765888 0.000000
10 O 2 Z 0.000000 0.000000 0.000000 0.000000 0.934639
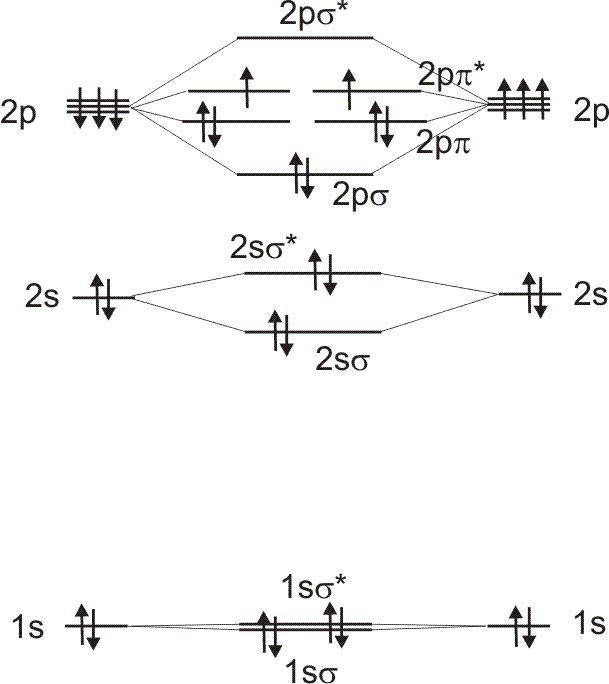 |
Links ist ein einfaches MO-Schema des Sauerstoffmoleküls
dargestellt. Der Vergleich mit den obigen MO's ergibt: 1s_sigma und 1s_sigma* entsprechen den beiden MO's A1g und A2u*. Ihre Energieaufspaltung ist minimal, d.h. es sind faktisch die 1s-AO's. |
2s_sigma und 2s_sigma* entsprechen dem 3. und 4.MO A1g und A2u. Beide liegen
energetisch wesentlich höher als die 1s_sigma MO's. Ihre Aufspaltung beträgt
0.5 eV. Das nächste MO ist ein bindendes sigma-MO, welches durch die
Kombination der 2pz-AO's zustande kommt (A1g). Es liegt ca. 0.1 eV über
2s_sigma*. Es folgen die zwei entarteten bindenden pi-MO's (Eu, ca. 0.1 eV
über dem vorhergehenden A1g-MO). Die beiden antibindenden pi-MO's (Eg) liegen
0.76 eV höher. Das antibindende 2p_sigma MO (A2u) liegt 0.89 eV höher. Man
erkennt aber, dass das MO-Schema qualitative mit den berechneten MO-Energien
übereinstimmt (für die beta-Spins). Beim Singulettsauerstoff
(beide Elektronen befinden sich in einem ppi-MO) stimmt diese MO-Reihenfolge bereits
nicht mehr.
*) Bei der Besetzung durch alpha-Spins ist das energetisch niedrigste MO A2u.
Das hängt damit zusammen, dass definitionsgemäß die alpha-Spins in der
Überzahl sind und damit die Elektronenabstoßung größer ist.
------------------------------
properties for the UHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -261.4945970652
TWO ELECTRON ENERGY = 84.6268793390
NUCLEAR REPULSION ENERGY = 27.8155162052
------------------
TOTAL ENERGY = -149.0522015211
ELECTRON-ELECTRON POTENTIAL ENERGY = 84.6268793390
NUCLEUS-ELECTRON POTENTIAL ENERGY = -410.1165497786
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 27.8155162052
------------------
TOTAL POTENTIAL ENERGY = -297.6741542345
TOTAL KINETIC ENERGY = 148.6219527134
VIRIAL RATIO (V/T) = 2.0028949210
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -92.2408383477
BARE H ENERGY= -261.4945970652
ELECTRONIC ENERGY = -176.8677177064
KINETIC ENERGY= 148.6219527134
N-N REPULSION= 27.8155162052
TOTAL ENERGY= -149.0522015012
SIGMA PART(1+2)= -146.0015961861
(K,V1,2)= 133.7933843767 -336.7355357650 56.9405552022
PI PART(1+2)= -30.8661215204
(K,V1,2)= 14.8285683367 -73.3810140136 27.6863241566
SIGMA SKELETON, ERROR= -118.1860799809 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
ALPHA ORBITALS
1
2 3
4 5
1.000000 1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000 0.500000
6
7 8
9
1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000
In 9 MO's befinden sich je ein alpha-Spin Elektron gleichmäßig verteilt auf beide O-Atome.
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
BETA ORBITALS
1
2 3
4 5
1.000000 1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000 0.500000
6 7
1.000000 1.000000
1 0.500000 0.500000
2 0.500000 0.500000
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 O1 1.000000 1.000000
2 O1 1.000000 1.000000
Beide Atome enthalten je ein ungepaartes Elektron.
********* ALL ELECTRONS ********
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99930 1.99826
2 O 1 S 1.89213 1.85065
3 O 1 X 1.50000 1.50000
4 O 1 Y 1.50000 1.50000
5 O 1 Z 1.10857 1.15109
6 O 2 S 1.99930 1.99826
7 O 2 S 1.89213 1.85065
8 O 2 X 1.50000 1.50000
9 O 2 Y 1.50000 1.50000
10 O 2 Z 1.10857 1.15109
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1
2
1 7.7498536
2 0.2501464 7.7498536
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O1 8.000000 0.000000 8.000000 0.000000
2 O1 8.000000 0.000000 8.000000 0.000000
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
ATOM PAIR DIST ORDER
1 2 1.218 2.000
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O1 2.502 2.000 0.502
2 O1 2.502 2.000 0.502
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 O1 8.0 0.2079407
2 O1 8.0 0.2079407
TOTAL NUMBER OF SHELLS = 10
TOTAL NUMBER OF BASIS FUNCTIONS = 18
NUMBER OF ELECTRONS = 16
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 3
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 9
NUMBER OF OCCUPIED ORBITALS (BETA ) = 7
TOTAL NUMBER OF ATOMS = 2
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
O
O
1 O 0.0000000 1.2073057 *
2 O 1.2073057 * 0.0000000
------------------
MOLECULAR ORBITALS
------------------
**** ALPHA SET ****
1 2
3 4
5
-20.7605 -20.7598 -1.7610 -1.2029 -0.8570
A1G A2U
A1G A2U EU
6 7
8 9 10
-0.8570
-0.7544 -0.5715 -0.5715 0.3561
EU
A1G EG
EG A2U
A1g schiebt sich zwischen die pi- und die pi*-MO's.
1 O 1 S 0.000000 0.056853 0.000000 0.000000 -0.097547
2 O 1 S 0.000000 -0.110686 0.000000 0.000000 0.181490
3 O 1 S 0.000000 -0.242554 0.000000 0.000000 1.302433
4 O 1 X 0.000000 0.000000 0.000000 0.627054 0.000000
5 O 1 Y 0.552006 0.000000 0.627054 0.000000 0.000000
6 O 1 Z 0.000000 0.538713 0.000000 0.000000 0.436875
7 O 1 X 0.000000 0.000000 0.000000 0.281542 0.000000
8 O 1 Y 0.163042 0.000000 0.281542 0.000000 0.000000
9 O 1 Z 0.000000 0.144437 0.000000 0.000000 1.168677
10 O 2 S 0.000000 0.056853 0.000000 0.000000 0.097547
11 O 2 S 0.000000 -0.110686 0.000000 0.000000 -0.181490
12 O 2 S 0.000000 -0.242554 0.000000 0.000000 -1.302433
13 O 2 X 0.000000 0.000000 0.000000 -0.627054 0.000000
14 O 2 Y 0.552006 0.000000 -0.627054 0.000000 0.000000
15 O 2 Z 0.000000 -0.538713 0.000000 0.000000 0.436875
16 O 2 X 0.000000 0.000000 0.000000 -0.281542 0.000000
17 O 2 Y 0.163042 0.000000 -0.281542 0.000000 0.000000
18 O 2 Z 0.000000 -0.144437 0.000000 0.000000 1.168677
Beta set
6 7 8 9 10
-0.5785 -0.5785 0.0890 0.0890 0.4192
EU EU EG EG A2U
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -261.3246246618
TWO ELECTRON ENERGY = 83.6831551380
NUCLEAR REPULSION ENERGY = 28.0520033221
------------------
TOTAL ENERGY = -149.5894662016
ELECTRON-ELECTRON POTENTIAL ENERGY = 83.6831551380
NUCLEUS-ELECTRON POTENTIAL ENERGY = -411.1069088367
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 28.0520033221
------------------
TOTAL POTENTIAL ENERGY = -299.3717503765
TOTAL KINETIC ENERGY = 149.7822841749
VIRIAL RATIO (V/T) = 1.9987126784
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -93.9583153401
BARE H ENERGY= -261.3246246618
ELECTRONIC ENERGY = -177.6414700009
KINETIC ENERGY= 149.7822841749
N-N REPULSION= 28.0520033221
TOTAL ENERGY= -149.5894666788
SIGMA PART(1+2)= -146.8228115320
(K,V1,2)= 134.7090675941 -338.4103956615 56.8785165355
PI PART(1+2)= -30.8186584689
(K,V1,2)= 15.0732165809 -72.6965131752 26.8046381253
SIGMA SKELETON, ERROR= -118.7708082098 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
ALPHA ORBITALS
1 2 3 4 5
1.000000 1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000 0.500000
6 7 8 9
1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
BETA ORBITALS
1 2 3 4 5
1.000000 1.000000 1.000000 1.000000 1.000000
1 0.500000 0.500000 0.500000 0.500000 0.500000
2 0.500000 0.500000 0.500000 0.500000 0.500000
6 7
1.000000 1.000000
1 0.500000 0.500000
2 0.500000 0.500000
ATOMIC SPIN POPULATION (ALPHA MINUS BETA)
ATOM MULL.POP. LOW.POP.
1 O 1.000000 1.000000
2 O 1.000000 1.000000
********* ALL ELECTRONS ********
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 2.00008 1.99474
2 O 1 S 1.00601 0.94323
3 O 1 S 0.91730 0.79410
4 O 1 X 1.12866 1.06676
5 O 1 Y 1.12866 1.06676
6 O 1 Z 0.95731 0.91096
7 O 1 X 0.37134 0.43324
8 O 1 Y 0.37134 0.43324
9 O 1 Z 0.11930 0.35697
10 O 2 S 2.00008 1.99474
11 O 2 S 1.00601 0.94323
12 O 2 S 0.91730 0.79410
13 O 2 X 1.12866 1.06676
14 O 2 Y 1.12866 1.06676
15 O 2 Z 0.95731 0.91096
16 O 2 X 0.37134 0.43324
17 O 2 Y 0.37134 0.43324
18 O 2 Z 0.11930 0.35697
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2
1 7.9347257
2 0.0652743 7.9347257
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O 8.000000 0.000000 8.000000 0.000000
2 O 8.000000 0.000000 8.000000 0.000000
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
ATOM PAIR DIST ORDER
1 2 1.207 1.723
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O 2.315 1.723 0.593
2 O 2.315 1.723 0.593
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 O 8.0 0.4862139
2 O 8.0 0.4862139
GBASIS=TZV NDFUNC=1
GBASIS=TZV NDFUNC=3 NFFUNC=1 DIFFSP=.TRUE.
NFFUNC=1: f-type polarization function
DIFFSP=.TRUE.: diffuse sp-type function
BASIS OPTIONS
-------------
GBASIS=TZV IGAUSS= 0 POLAR=HONDO7
NDFUNC= 3 DIFFSP= T
NPFUNC= 0 DIFFS= F
SPLIT3= 4.00000000 1.00000000 0.25000000
TOTAL NUMBER OF BASIS FUNCTIONS = 92
SPIN SZ =1.000
S-SQUARED =2.043
------------------
MOLECULAR ORBITALS
------------------
**** ALPHA SET ****
1 2 3 4 5
-20.7463 -20.7454 -1.7599 -1.1889 -0.8627
A1G A2U A1G A2U EU
6 7 8 9 10
-0.8627 -0.7831 -0.5374 -0.5374 0.1455
EU A1G EG EG A2U
11 12 13 14 15
0.1540 0.1810 0.1810 0.2474 0.2474
A1G EU EU EG EG
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -263.9186639420
TWO ELECTRON ENERGY = 84.9896289498
NUCLEAR REPULSION ENERGY = 29.2403127593
------------------
TOTAL ENERGY = -149.6887222329
ELECTRON-ELECTRON POTENTIAL ENERGY = 84.9896289498
NUCLEUS-ELECTRON POTENTIAL ENERGY = -413.5950018397
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 29.2403127593
------------------
TOTAL POTENTIAL ENERGY = -299.3650601306
TOTAL KINETIC ENERGY = 149.6763378977
VIRIAL RATIO (V/T) = 2.0000827408
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.06614 1.03839
2 O 1 S 0.86309 0.79489
3 O 1 S 0.25152 0.23732
4 O 1 S 0.77288 0.31461
5 O 1 S 0.91427 0.25895
6 O 1 X 0.69161 0.51524
7 O 1 Y 0.69161 0.51524
8 O 1 Z 0.59550 0.42563
9 O 1 X 0.58278 0.34415
10 O 1 Y 0.58278 0.34415
11 O 1 Z 0.41817 0.32865
12 O 1 X 0.18963 0.16063
13 O 1 Y 0.18963 0.16063
14 O 1 Z 0.07093 0.14733
15 O 1 S 0.04642 0.08317
16 O 1 X 0.00465 0.04427
17 O 1 Y 0.00465 0.04427
18 O 1 Z -0.00565 0.04499
19 O 1 XX -0.00852 0.06351
20 O 1 YY -0.00852 0.06351
21 O 1 ZZ -0.00843 0.06427
22 O 1 XY 0.00000 0.00000
23 O 1 XZ 0.00013 0.00021
24 O 1 YZ 0.00013 0.00021
25 O 1 XX 0.02194 0.18865
26 O 1 YY 0.02194 0.18865
27 O 1 ZZ 0.05252 0.19589
28 O 1 XY 0.00000 0.00000
29 O 1 XZ 0.00801 0.00721
30 O 1 YZ 0.00801 0.00721
31 O 1 XX -0.03225 0.03972
32 O 1 YY -0.03225 0.03972
33 O 1 ZZ -0.02488 0.12927
34 O 1 XY 0.00000 0.00000
35 O 1 XZ 0.01655 0.07163
36 O 1 YZ 0.01655 0.07163
37 O XXX -0.00158 0.18008
38 O YYY -0.00158 0.18008
39 O ZZZ 0.02624 0.21005
40 O XXY -0.00053 0.08349
41 O XXZ -0.00053 0.07142
42 O YYX -0.00053 0.08349
43 O YYZ -0.00053 0.07142
44 O ZZX 0.00875 0.09309
45 O ZZY 0.00875 0.09309
46 O XYZ 0.00000 0.00000
47 O 2 S 1.06614 1.03839
48 O 2 S 0.86309 0.79489
49 O 2 S 0.25152 0.23732
50 O 2 S 0.77288 0.31461
51 O 2 S 0.91427 0.25895
52 O 2 X 0.69161 0.51524
53 O 2 Y 0.69161 0.51524
54 O 2 Z 0.59550 0.42563
55 O 2 X 0.58278 0.34415
56 O 2 Y 0.58278 0.34415
57 O 2 Z 0.41817 0.32865
58 O 2 X 0.18963 0.16063
59 O 2 Y 0.18963 0.16063
60 O 2 Z 0.07093 0.14733
61 O 2 S 0.04642 0.08317
62 O 2 X 0.00465 0.04427
63 O 2 Y 0.00465 0.04427
64 O 2 Z -0.00565 0.04499
65 O 2 XX -0.00852 0.06351
66 O 2 YY -0.00852 0.06351
67 O 2 ZZ -0.00843 0.06427
68 O 2 XY 0.00000 0.00000
69 O 2 XZ 0.00013 0.00021
70 O 2 YZ 0.00013 0.00021
71 O 2 XX 0.02194 0.18865
72 O 2 YY 0.02194 0.18865
73 O 2 ZZ 0.05252 0.19589
74 O 2 XY 0.00000 0.00000
75 O 2 XZ 0.00801 0.00721
76 O 2 YZ 0.00801 0.00721
77 O 2 XX -0.03225 0.03972
78 O 2 YY -0.03225 0.03972
79 O 2 ZZ -0.02488 0.12927
80 O 2 XY 0.00000 0.00000
81 O 2 XZ 0.01655 0.07163
82 O 2 YZ 0.01655 0.07163
83 O XXX -0.00158 0.18008
84 O YYY -0.00158 0.18008
85 O ZZZ 0.02624 0.21005
86 O XXY -0.00053 0.08349
87 O XXZ -0.00053 0.07142
88 O YYX -0.00053 0.08349
89 O YYZ -0.00053 0.07142
90 O ZZX 0.00875 0.09309
91 O ZZY 0.00875 0.09309
92 O XYZ 0.00000 0.00000
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
ATOM PAIR DIST BOND ORDER
1 2 1.158
1.810
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O 2.446 1.810 0.636
2 O 2.446 1.810 0.636
ATOMIC SPIN DENSITY AT THE NUCLEUS (A.U.)
-----------------------------------------
1 O 8.0 0.3496517
2 O 8.0 0.3496517
CPU TIME: STEP = 0.00 , TOTAL = 73.9 SECONDS ( 1.2 MIN)
-----------------
MCSCF CALCULATION
-----------------
GROUP=D2H NPRT= 0
FORS= T INTACT= F
FOCI= F MXNINT= 20000
SOCI= F MXNEME= 7500
IEXCIT= 0 NWORD = 180018
-CORE- -INTERNAL- -EXTERNAL-
NFZC= 0 NDOC= 7 NEXT= 0
NMCC= 0 NAOS= 0 NFZV= 0
NBOS= 0
NALP= 2
NVAL= 1
THE MAXIMUM ELECTRON EXCITATION WILL BE 4
SYMMETRIES FOR THE 0 CORE, 10 ACTIVE, 0
EXTERNAL MO-S ARE
ACTIVE= AG B1U AG B1U B3U B2U AG
B2G B3G B1U
DOC DOC DOC DOC DOC DOC DOC ALP
ALP VAL
-----------------------
-MCHF- NATURAL ORBITALS
-----------------------
1 2 3 4 5
2.0000
2.0000 1.9990 1.9956 1.9401
1 O 1 S 0.710733 0.708038 -0.146102 -0.165259 0.000000
2 O 1 S -0.015676 -0.003742 0.659086 0.781154 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.667686
4 O 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 O 1 Z -0.002507 -0.005373 0.040321 0.125385 0.000000
6 O 2 S 0.710733 -0.708038 -0.146102 0.165259 0.000000
7 O 2 S -0.015676 0.003742 0.659086 -0.781154 0.000000
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.667686
9 O 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 O 2 Z 0.002507 -0.005373 -0.040321 0.125385 0.000000
d.h. man hätte die ersten vier MO's einfrieren können
6 7
8 9
10
1.9401
1.9428 1.0591 1.0591
0.0641
1 O 1 S 0.000000 -0.005353 0.000000 0.000000 0.074413
2 O 1 S 0.000000 0.105164 0.000000 0.000000 -0.457758
3 O 1 X 0.000000 0.000000 0.754450 0.000000
0.000000
4 O 1 Y 0.667686 0.000000 0.000000 0.754450
0.000000
5 O 1 Z 0.000000 0.637367 0.000000 0.000000
0.899010
6 O 2 S 0.000000 -0.005353 0.000000 0.000000 -0.074413
7 O 2 S 0.000000 0.105164 0.000000 0.000000
0.457758
8 O 2 X 0.000000 0.000000 -0.754450 0.000000 0.000000
9 O 2 Y 0.667686 0.000000 0.000000 -0.754450 0.000000
10 O 2 Z 0.000000 -0.637367 0.000000 0.000000 0.899010
Die MO's 8 und 9 werden durch
jeweils ein Elektron besetzt.
-------------------------
-MCHF- OPTIMIZED ORBITALS
-------------------------
1 2 3 4 5
0.0000 0.0000 0.0000 0.0000 0.0000
AG B1U AG B1U B3U
1 O 1 S 0.411822 0.545166 0.597013 -0.478627 0.000000
2 O 1 S 0.438907 0.393869 -0.477793 0.726438 0.000000
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.667686
4 O 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 O 1 Z -0.005685 -0.029496 0.009147 0.045871 0.000000
6 O 2 S 0.411822 -0.545166 0.597013 0.478627 0.000000
7 O 2 S 0.438907 -0.393869 -0.477793 -0.726438 0.000000
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.667686
9 O 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 O 2 Z 0.005685 -0.029496 -0.009147 0.045871 0.000000
6 7 8 9 10
0.0000 0.0000 0.0000 0.0000 0.0000
B2U AG B2G B3G B1U
1 O 1 S 0.000000 -0.022244 0.000000 0.000000 0.088743
2 O 1 S 0.000000 0.157398 0.000000 0.000000 -0.370017
3 O 1 X 0.000000 0.000000 0.754450 0.000000 0.000000
4 O 1 Y 0.667686 0.000000 0.000000 0.754450 0.000000
5 O 1 Z 0.000000 0.638555 0.000000 0.000000 0.906088
6 O 2 S 0.000000 -0.022244 0.000000 0.000000 -0.088743
7 O 2 S 0.000000 0.157398 0.000000 0.000000 0.370017
8 O 2 X 0.000000 0.000000 -0.754450 0.000000 0.000000
9 O 2 Y 0.667686 0.000000 0.000000 -0.754450 0.000000
10 O 2 Z 0.000000 -0.638555 0.000000 0.000000 0.906088
STATE # 1 ENERGY = -149.175638526
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
1 -0.192366 2222112220
2 0.941451 2222222110
8 -0.121308 2222121211
19 0.121308 2222211121
29 0.101502 2221222111
32 0.122637 2122222111
38 0.056144 2222110222
95 -0.089288 2222220112
--------------------------------
properties for the MCSCF density
--------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -258.6160540848
TWO ELECTRON ENERGY = 83.2445218906
NUCLEAR REPULSION ENERGY = 26.1958936686
------------------
TOTAL ENERGY = -149.1756385256
ELECTRON-ELECTRON POTENTIAL ENERGY = 83.2445218906
NUCLEUS-ELECTRON POTENTIAL ENERGY = -407.4303388171
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 26.1958936686
------------------
TOTAL POTENTIAL ENERGY = -297.9899232579
TOTAL KINETIC ENERGY = 148.8142847323
VIRIAL RATIO (V/T) = 2.0024282198
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
1.999999 1.999999 1.999034 1.995641 1.940099
1 1.000000 1.000000 0.999517 0.997820 0.970050
2 1.000000 1.000000 0.999517 0.997820 0.970050
6 7 8 9 10
1.940099 1.942819 1.059103 1.059103 0.064103
1 0.970050 0.971410 0.529552 0.529552 0.032052
2 0.970050 0.971410 0.529552 0.529552 0.032052
WARNING! MCSCF POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR MCSCF FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY MCSCF WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99951 1.99863
2 O 1 S 1.92529 1.89460
3 O 1 X 1.49960 1.49960
4 O 1 Y 1.49960 1.49960
5 O 1 Z 1.07601 1.10757
6 O 2 S 1.99951 1.99863
7 O 2 S 1.92529 1.89460
8 O 2 X 1.49960 1.49960
9 O 2 Y 1.49960 1.49960
10 O 2 Z 1.07601 1.10757
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2
1 7.8209333
2 0.1790667 7.8209333
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O 8.000000 0.000000 8.000000 0.000000
2 O 8.000000 0.000000 8.000000 0.000000
STO-6G
TOTAL NUMBER OF SHELLS = 4
TOTAL NUMBER OF BASIS FUNCTIONS = 10
NUMBER OF ELECTRONS = 16
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 8
NUMBER OF OCCUPIED ORBITALS (BETA ) = 8
TOTAL NUMBER OF ATOMS = 2
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
O2-Molekuel
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
O O
1 O 0.0000000 1.2708592 *
2 O 1.2708592 * 0.0000000
| STO-6G | DZV | ||||||
| R /Å | 1.2709 | 1.270 |
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-20.6790 -20.6789 -1.5051 -1.0215 -0.5686
A1G A2U A1G A2U A1G
1 O 1 S 0.705204 0.704809 -0.161156 -0.177040 0.057267
2 O 1 S 0.007394 0.011071 0.593189 0.775892 -0.302074
3 O 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 O 1 Y 0.000000 0.000000 0.000000 0.000000 0.000000
5 O 1 Z 0.000236 0.003270 0.153580 -0.142310 0.619262
6 O 2 S 0.705204 -0.704809 -0.161156 0.177040 0.057267
7 O 2 S 0.007394 -0.011071 0.593189 -0.775892 -0.302074
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y 0.000000 0.000000 0.000000 0.000000 0.000000
10 O 2 Z -0.000236 0.003270 -0.153580 -0.142310 -0.619262
6 7
8 9
10
-0.5525 -0.5525
-0.0997 -0.0997 0.6134
EU
EU EG
EG A2U
1 O 1 S 0.000000 0.000000 0.000000 0.000000 -0.079386
2 O 1 S 0.000000 0.000000 0.000000 0.000000 0.493958
3 O 1 X 0.665566 0.000000 0.757543 0.000000 0.000000
4 O 1 Y 0.000000 0.665566 0.000000 0.757543 0.000000
5 O 1 Z 0.000000 0.000000 0.000000 0.000000 0.908740
6 O 2 S 0.000000 0.000000 0.000000 0.000000 0.079386
7 O 2 S 0.000000 0.000000 0.000000 0.000000 -0.493958
8 O 2 X 0.665566 0.000000 -0.757543 0.000000 0.000000
9 O 2 Y 0.000000 0.665566 0.000000 -0.757543 0.000000
10 O 2 Z 0.000000 0.000000 0.000000 0.000000 0.908740
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -259.3999431588
TWO ELECTRON ENERGY = 84.0421532590
NUCLEAR REPULSION ENERGY = 26.6491698424
------------------
TOTAL ENERGY = -148.7086200574
ELECTRON-ELECTRON POTENTIAL ENERGY = 84.0421532590
NUCLEUS-ELECTRON POTENTIAL ENERGY = -407.9845155001
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 26.6491698424
------------------
TOTAL POTENTIAL ENERGY = -297.2931923988
TOTAL KINETIC ENERGY = 148.5845723414
VIRIAL RATIO (V/T) = 2.0008348627
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -91.3156366408
BARE H ENERGY= -259.3999431588
ELECTRONIC ENERGY = -175.3577898998
KINETIC ENERGY= 148.5845723414
N-N REPULSION= 26.6491698424
TOTAL ENERGY= -148.7086200574
SIGMA PART(1+2)= -145.2497973771
(K,V1,2)= 133.7512249240 -335.3447439836 56.3437216826
PI PART(1+2)= -30.1079925227
(K,V1,2)= 14.8333474174 -72.6397715165 27.6984315763
SIGMA SKELETON, ERROR= -118.6006275346 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2
3 4
5
2.000000 2.000000 2.000000 2.000000 2.000000
1 1.000000 1.000000 1.000000 1.000000 1.000000
2 1.000000 1.000000 1.000000 1.000000 1.000000
6 7
8
2.000000 2.000000 2.000000
1 1.000000 1.000000 1.000000
2 1.000000 1.000000 1.000000
Man sieht, dass 2 Elektronen das MO Nr.8 Eg,
pi*) besetzen, wie für
![]() erwartet.
erwartet.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 O 1 S 1.99940 1.99842
2 O 1 S 1.91016 1.87656
3 O 1 X 2.00000 2.00000
4 O 1 Y 1.00000 1.00000
5 O 1 Z 1.09043 1.12502
6 O 2 S 1.99940 1.99842
7 O 2 S 1.91016 1.87656
8 O 2 X 2.00000 2.00000
9 O 2 Y 1.00000 1.00000
10 O 2 Z 1.09043 1.12502
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2
1 7.7549166
2 0.2450834 7.7549166
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 O 8.000000 0.000000 8.000000 0.000000
2 O 8.000000 0.000000 8.000000 0.000000
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.271 2.000
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O 2.000 2.000 0.000
2 O 2.000 2.000 0.000
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -132.1176760719
TWO ELECTRON ENERGY = 41.5139974873
NUCLEAR REPULSION ENERGY = 15.4409588781
------------------
TOTAL ENERGY = -75.1627197064
ELECTRON-ELECTRON POTENTIAL ENERGY = 41.5139974873
NUCLEUS-ELECTRON POTENTIAL ENERGY = -208.4509254972
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 15.4409588781
------------------
TOTAL POTENTIAL ENERGY = -151.4959691318
TOTAL KINETIC ENERGY = 76.3332494253
VIRIAL RATIO (V/T) = 1.9846655327
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -49.0896810388
BARE H ENERGY= -132.1176760719
ELECTRONIC ENERGY = -90.6036785553
KINETIC ENERGY= 76.3332494253
N-N REPULSION= 15.4409588781
TOTAL ENERGY= -75.1627196772
SIGMA PART(1+2)= -77.6776232988
(K,V1,2)= 71.3129525301 -179.0816419726 30.0910661438
PI PART(1+2)= -12.9260552566
(K,V1,2)= 5.0202968952 -29.3692835246 11.4229313728
SIGMA SKELETON, ERROR= -62.2366644206 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
Total Energy, triplet-singlet splitting
| Triplet | Singlet | Diff | |
| STO-6G | -75.2279 | -75.3575 | |
| DZV | -75.4226 | -75.1627 | |
| TZV | -75.4395 | ||
| TZV+ND=1 | -75.4711 | ||
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99849 1.99732
2 C 1 S 1.58220 1.46546
3 C 1 X 1.00000 1.00000
4 C 1 Y 1.00000 1.00000
5 C 1 Z 0.41932 0.53722
6 C 2 S 1.99849 1.99732
7 C 2 S 1.58220 1.46546
8 C 2 X 1.00000 1.00000
9 C 2 Y 1.00000 1.00000
10 C 2 Z 0.41932 0.53722
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.234 3.333
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 O 3.333 3.333 0.000
2 O 3.333 3.333 0.000