
| Arbeiten mit GAMESS |
Der Bindungsausgleich bei Polymethinen hat seine eigentliche Ursache in der Ungeradzahligkeit der p-Zentrenkette. Er ist aber nicht auf p-Orbitale beschränkt (s. ungeradzahlige lineare Kette von H-Atomen). Die Stammverbindung der Polymethine ist das Allyl-Anion:
Die beiden Resonanzstrukturen suggerieren den Bindungsausgleich.
Als stabile Verbindung (allerdings mit einer Methylgruppe anstelle der H-Atome) wird das obige 4p-Elektronensystem durch folgendes Kation realisiert (jeweils ein Elektronenpaar durch die beiden N-Atome, ein p-Elektron durch das zentrale C-Atom, minus ein Elektron wegen der positiven Ladung):
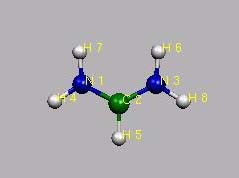
Interessant ist, dass mit zunehmender Zahl an C-Atomen (ungeradzahlig, also 1, 3, 5 ,7, ..) die Wellenlänge der langwelligsten Absorptionsbande nahezu linear zunimmt:
| C-Atome | l / nm | lg e |
| 1 | 224 | 4.16 |
| 3 | 312.5 | 4.81 |
| 5 | 416 | 5.08 |
| 7 | 519 | 5.32 |
| 9 | 625 | 5.47 |
| 11 | 734.5 | 5.55 |
| 13 | 848 | 5.34 |
Basisatz: STO-6G, 31G, 311G,
Input
!
! Polymethin, 4 pi-Elektronen
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=OPTIMIZE COORD=ZMTMPC $END
$SYSTEM TIMLIM=10 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Polymethin, 4 pi-Elektronen
C1
N 00000.0000 0 00000.0000 0 00000.0000 0 0 0 0
C 00001.3488 1 00000.0000 0 00000.0000 0 1 0 0
N 00001.3487 1 00122.5834 1 00000.0000 0 2 1 0
H 00000.9903 1 00119.4361 1 00179.9999 1 1 2 3
H 00001.1111 1 00118.7020 1 00180.0000 1 2 1 3
H 00000.9917 1 00122.3903 1 00000.0000 1 3 2 1
H 00000.9919 1 00122.3841 1 00179.9999 1 1 2 5
H 00000.9901 1 00119.3931 1 00000.0000 1 3 2 5
$END
Die Geometrie-Eingabe resultiert aus dem Molekül-Builder
von "Hyperchem", speichern als MOPAC-Typ-Z-Matrix.
Output
YOUR FULLY SUBSTITUTED Z-MATRIX IS
N
C 1 1.3488000
N 2 1.3487000 1 122.5834
H 1 0.9903000 2 119.4361 3 179.9999 0
H 2 1.1111000 1 118.7020 3 180.0000 0
H 3 0.9917000 2 122.3903 1 0.0000 0
H 1 0.9919000 2 122.3841 5 179.9999 0
H 3 0.9901000 2 119.3931 5 0.0000 0
THE MOMENTS OF INERTIA ARE (AMU-ANGSTROM**2)
IXX= 9.037 IYY= 50.580 IZZ= 59.616
ATOM ATOMIC COORDINATES (BOHR)
CHARGE X Y Z
N 7.0 -2.2354819182 0.3609787725 0.0000000000
C 6.0 0.0001275633 -0.8632556179 0.0000000000
N 7.0 2.2354474339 0.3611141702 0.0000000000
H 1.0 -3.8249597323 -0.6267873642 0.0000000000
H 1.0 0.0000029082 -2.9629301592 0.0000000000
H 1.0 2.3557015096 2.2312932015 0.0000000000
H 1.0 -2.3557470838 2.2315358164 0.0000000000
H 1.0 3.8239626593 -0.6274841375 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N C N H
1 N 0.0000000 1.3488000 * 2.3659141 * 0.9903000 *
2 C 1.3488000 * 0.0000000 1.3487000 * 2.0280134 *
3 N 2.3659141 * 1.3487000 * 0.0000000 3.2493588
4 H 0.9903000 * 2.0280134 * 3.2493588 0.0000000
5 H 2.1197339 * 1.1111000 * 2.1197815 * 2.3717475 *
6 H 2.6234088 * 2.0580139 * 0.9917000 * 3.6034289
7 H 0.9919000 * 2.0582124 * 2.6234356 * 1.7006770 *
8 H 3.2489039 2.0273293 * 0.9901000 * 4.0476357
H H H H
1 N 2.1197339 * 2.6234088 * 0.9919000 * 3.2489039
2 C 1.1111000 * 2.0580139 * 2.0582124 * 2.0273293 *
3 N 2.1197815 * 0.9917000 * 2.6234356 * 0.9901000 *
4 H 2.3717475 * 3.6034289 1.7006770 * 4.0476357
5 H 0.0000000 3.0181327 3.0182609 2.3711024 *
6 H 3.0181327 0.0000000 2.4931914 * 1.7006606 *
7 H 3.0182609 2.4931914 * 0.0000000 3.6031806
8 H 2.3711024 * 1.7006606 * 3.6031806 0.0000000
* ... LESS THAN 3.000
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
N
1 S 1 1027.828458 1.185541 ( 0.009164)
1 S 2 188.451223 1.789359 ( 0.049361)
1 S 3 52.721861 2.350183 ( 0.168538)
1 S 4 18.111382 2.318652 ( 0.370563)
1 S 5 7.033180 1.281976 ( 0.416492)
1 S 6 2.896652 0.206248 ( 0.130334)
2 L 7 39.198808 -0.147969 ( -0.013253) 0.525635 ( 0.003760)
2 L 8 7.758467 -0.155691 ( -0.046992) 0.695446 ( 0.037679)
2 L 9 2.411326 -0.046594 ( -0.033785) 0.744820 ( 0.173897)
2 L 10 0.927724 0.168591 ( 0.250242) 0.542535 ( 0.418036)
2 L 11 0.402911 0.214496 ( 0.595117) 0.194858 ( 0.425860)
2 L 12 0.184684 0.048330 ( 0.240706) 0.017552 ( 0.101708)
C
3 S 13 742.737049 0.929185 ( 0.009164)
3 S 14 136.180025 1.402437 ( 0.049361)
3 S 15 38.098264 1.841991 ( 0.168538)
3 S 16 13.087782 1.817278 ( 0.370563)
3 S 17 5.082369 1.004768 ( 0.416492)
3 S 18 2.093200 0.161650 ( 0.130334)
4 L 19 30.497240 -0.122578 ( -0.013253) 0.384077 ( 0.003760)
4 L 20 6.036200 -0.128975 ( -0.046992) 0.508157 ( 0.037679)
4 L 21 1.876046 -0.038599 ( -0.033785) 0.544234 ( 0.173897)
4 L 22 0.721783 0.139661 ( 0.250242) 0.396426 ( 0.418036)
4 L 23 0.313471 0.177689 ( 0.595117) 0.142381 ( 0.425860)
4 L 24 0.143687 0.040037 ( 0.240706) 0.012825 ( 0.101708)
N
5 S 25 1027.828458 1.185541 ( 0.009164)
5 S 26 188.451223 1.789359 ( 0.049361)
5 S 27 52.721861 2.350183 ( 0.168538)
5 S 28 18.111382 2.318652 ( 0.370563)
5 S 29 7.033180 1.281976 ( 0.416492)
5 S 30 2.896652 0.206248 ( 0.130334)
6 L 31 39.198808 -0.147969 ( -0.013253) 0.525635 ( 0.003760)
6 L 32 7.758467 -0.155691 ( -0.046992) 0.695446 ( 0.037679)
6 L 33 2.411326 -0.046594 ( -0.033785) 0.744820 ( 0.173897)
6 L 34 0.927724 0.168591 ( 0.250242) 0.542535 ( 0.418036)
6 L 35 0.402911 0.214496 ( 0.595117) 0.194858 ( 0.425860)
6 L 36 0.184684 0.048330 ( 0.240706) 0.017552 ( 0.101708)
H
7 S 37 35.523221 0.095030 ( 0.009164)
7 S 38 6.513144 0.143430 ( 0.049361)
7 S 39 1.822143 0.188385 ( 0.168538)
7 S 40 0.625955 0.185857 ( 0.370563)
7 S 41 0.243077 0.102760 ( 0.416492)
7 S 42 0.100112 0.016532 ( 0.130334)
H
8 S 43 35.523221 0.095030 ( 0.009164)
8 S 44 6.513144 0.143430 ( 0.049361)
8 S 45 1.822143 0.188385 ( 0.168538)
8 S 46 0.625955 0.185857 ( 0.370563)
8 S 47 0.243077 0.102760 ( 0.416492)
8 S 48 0.100112 0.016532 ( 0.130334)
H
9 S 49 35.523221 0.095030 ( 0.009164)
9 S 50 6.513144 0.143430 ( 0.049361)
9 S 51 1.822143 0.188385 ( 0.168538)
9 S 52 0.625955 0.185857 ( 0.370563)
9 S 53 0.243077 0.102760 ( 0.416492)
9 S 54 0.100112 0.016532 ( 0.130334)
H
10 S 55 35.523221 0.095030 ( 0.009164)
10 S 56 6.513144 0.143430 ( 0.049361)
10 S 57 1.822143 0.188385 ( 0.168538)
10 S 58 0.625955 0.185857 ( 0.370563)
10 S 59 0.243077 0.102760 ( 0.416492)
10 S 60 0.100112 0.016532 ( 0.130334)
H
11 S 61 35.523221 0.095030 ( 0.009164)
11 S 62 6.513144 0.143430 ( 0.049361)
11 S 63 1.822143 0.188385 ( 0.168538)
11 S 64 0.625955 0.185857 ( 0.370563)
11 S 65 0.243077 0.102760 ( 0.416492)
11 S 66 0.100112 0.016532 ( 0.130334)
TOTAL NUMBER OF SHELLS = 11
TOTAL NUMBER OF BASIS FUNCTIONS = 20
NUMBER OF ELECTRONS = 24
CHARGE OF MOLECULE = 1
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 12
NUMBER OF OCCUPIED ORBITALS (BETA ) = 12
TOTAL NUMBER OF ATOMS = 8
THE NUCLEAR REPULSION ENERGY IS 78.5981164488
NSERCH= 17 ENERGY= -149.0404398
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 N 7.0 0.0000129 0.0000091 0.0000000
2 C 6.0 0.0000111 0.0000015 0.0000000
3 N 7.0 0.0000017 0.0000014 0.0000000
4 H 1.0 0.0000242 -0.0000252 0.0000000
5 H 1.0 -0.0000059 0.0000147 0.0000000
6 H 1.0 -0.0000235 0.0000020 0.0000000
7 H 1.0 0.0000181 0.0000099 0.0000000
8 H 1.0 -0.0000386 -0.0000133 0.0000000
MAXIMUM GRADIENT = 0.0000386 RMS GRADIENT = 0.0000137
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Polymethin, 4 pi-Elektronen
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N 7.0 -1.1751131150 0.1799666365 0.0000000000
C 6.0 -0.0000726691 -0.4414077347 0.0000000000
N 7.0 1.1750279513 0.1798394988 0.0000000000
H 1.0 -2.0426627624 -0.3609470451 0.0000000000
H 1.0 -0.0001755854 -1.5413363041 0.0000000000
H 1.0 1.2520340097 1.2000736122 0.0000000000
H 1.0 -1.2520683582 1.2002120045 0.0000000000
H 1.0 2.0425295783 -0.3611203350 0.0000000000
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
N
C 1 1.3292201
N 2 1.3292139 1 124.2652956
H 1 1.0223649 2 120.1862475 3 180.0000000 0
H 2 1.0999286 1 117.8650198 3 180.0000000 0
H 3 1.0231361 2 122.1807564 1 0.0000000 0
H 1 1.0231435 2 122.1839285 5 180.0000000 0
H 3 1.0223486 2 120.1886859 5 0.0000000 0
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N C N H
1 N 0.0000000 1.3292201 * 2.3501411 * 1.0223649 *
2 C 1.3292201 * 0.0000000 1.3292139 * 2.0441742 *
3 N 2.3501411 * 1.3292139 * 0.0000000 3.2628184
4 H 1.0223649 * 2.0441742 * 3.2628184 0.0000000
5 H 2.0840734 * 1.0999286 * 2.0841184 * 2.3590406 *
6 H 2.6328049 * 2.0645174 * 1.0231361 * 3.6457938
7 H 1.0231435 * 2.0645601 * 2.6328609 * 1.7499306 *
8 H 3.2628208 2.0441795 * 1.0223486 * 4.0851923
H H H H
1 N 2.0840734 * 2.6328049 * 1.0231435 * 3.2628208
2 C 1.0999286 * 2.0645174 * 2.0645601 * 2.0441795 *
3 N 2.0841184 * 1.0231361 * 2.6328609 * 1.0223486 *
4 H 2.3590406 * 3.6457938 1.7499306 * 4.0851923
5 H 0.0000000 3.0138608 3.0138551 2.3591427 *
6 H 3.0138608 0.0000000 2.5041024 * 1.7499171 *
7 H 3.0138551 2.5041024 * 0.0000000 3.6458379
8 H 2.3591427 * 1.7499171 * 3.6458379 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 78.7058928763
ELECTRONIC ENERGY = -227.7463326315
TOTAL ENERGY = -149.0404397552
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-15.8827 -15.8826 -11.6415 -1.5074 -1.3961
A A A A A
1 N 1 S -0.599869 0.795189 0.000074 -0.134118 -0.155274
2 N 1 S -0.012045 0.016162 -0.003643 0.454152 0.545091
3 N 1 X -0.000437 0.000866 -0.002081 0.078875 0.014549
4 N 1 Y 0.000470 -0.000306 0.000934 -0.033120 -0.021751
5 N 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S 0.000064 0.000454 0.995644 -0.142195 -0.000005
7 C 2 S -0.000821 -0.005861 0.020713 0.354634 0.000014
8 C 2 X -0.004099 0.000574 0.000000 0.000009 -0.220256
9 C 2 Y -0.000312 -0.002231 0.000855 0.081289 0.000011
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 N 3 S 0.795201 0.599854 0.000074 -0.134126 0.155268
12 N 3 S 0.016021 0.012233 -0.003643 0.454180 -0.545070
13 N 3 X -0.000658 -0.000713 0.002081 -0.078881 0.014546
14 N 3 Y -0.000536 -0.000165 0.000934 -0.033110 0.021744
15 N 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 H 4 S 0.002343 -0.003072 -0.000308 0.055018 0.095522
17 H 5 S 0.000024 0.000168 -0.003898 0.046254 0.000003
18 H 6 S -0.003060 -0.002346 -0.000308 0.058232 -0.091170
19 H 7 S 0.002298 -0.003096 -0.000308 0.058227 0.091172
20 H 8 S -0.003097 -0.002310 -0.000308 0.055023 -0.095521
6 7 8 9 10
-1.0936 -0.9847 -0.9400 -0.8635 -0.8121
A A A A A
1 N 1 S 0.065425 0.014956 0.013141 -0.007159 -0.014815
2 N 1 S -0.266920 -0.072614 -0.075645 0.023870 0.064034
3 N 1 X 0.214796 0.322915 0.191060 0.424533 0.046402
4 N 1 Y -0.179909 0.216783 -0.391753 0.218151 -0.359767
5 N 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S -0.157238 -0.013514 0.000002 -0.000002 0.019066
7 C 2 S 0.513007 0.036087 -0.000006 0.000010 -0.090467
8 C 2 X -0.000008 0.000038 -0.376495 -0.247448 0.000027
9 C 2 Y -0.134580 0.356313 0.000021 0.000043 0.436412
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 N 3 S 0.065422 0.014958 -0.013139 0.007160 -0.014814
12 N 3 S -0.266905 -0.072625 0.075638 -0.023876 0.064027
13 N 3 X -0.214819 -0.322945 0.191090 0.424475 -0.046441
14 N 3 Y -0.179885 0.216823 0.391761 -0.218185 -0.359737
15 N 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 H 4 S -0.111891 -0.235831 0.003921 -0.317492 0.132827
17 H 5 S 0.219905 -0.176634 0.000005 -0.000016 -0.391017
18 H 6 S -0.164886 0.088605 0.265579 -0.131664 -0.263090
19 H 7 S -0.164893 0.088596 -0.265568 0.131655 -0.263105
20 H 8 S -0.111896 -0.235860 -0.003928 0.317467 0.132808
11 12 13 14 15
-0.7821 -0.6124 -0.0098 0.2701 0.3040
A A A A A
1 N 1 S 0.000000 0.000000 0.000000 -0.098368 -0.085849
2 N 1 S 0.000000 0.000000 0.000000 0.725530 0.617620
3 N 1 X 0.000000 0.000000 0.000000 -0.113737 0.045043
4 N 1 Y 0.000000 0.000000 0.000000 -0.264175 0.290265
5 N 1 Z 0.517742 0.712264 -0.519008 0.000000 0.000000
6 C 2 S 0.000000 0.000000 0.000000 0.033570 0.119746
7 C 2 S 0.000000 0.000000 0.000000 -0.185742 -0.776232
8 C 2 X 0.000000 0.000000 0.000000 0.000032 0.000007
9 C 2 Y 0.000000 0.000000 0.000000 -0.637061 0.331164
10 C 2 Z 0.495665 0.000007 0.919063 0.000000 0.000000
11 N 3 S 0.000000 0.000000 0.000000 -0.098337 -0.085854
12 N 3 S 0.000000 0.000000 0.000000 0.725302 0.617662
13 N 3 X 0.000000 0.000000 0.000000 0.113701 -0.045033
14 N 3 Y 0.000000 0.000000 0.000000 -0.264199 0.290252
15 N 3 Z 0.517760 -0.712251 -0.519009 0.000000 0.000000
16 H 4 S 0.000000 0.000000 0.000000 -0.722569 -0.127232
17 H 5 S 0.000000 0.000000 0.000000 -0.485656 0.821957
18 H 6 S 0.000000 0.000000 0.000000 -0.055153 -0.646786
19 H 7 S 0.000000 0.000000 0.000000 -0.055332 -0.646773
20 H 8 S 0.000000 0.000000 0.000000 -0.722461 -0.127253
16 17 18 19 20
0.3169 0.4155 0.4330 0.6132 0.6313
A A A A A
1 N 1 S 0.118283 0.021987 0.013241 0.027343 -0.037865
2 N 1 S -0.899312 -0.158369 -0.085881 -0.230874 0.341421
3 N 1 X -0.003250 0.642802 -0.459705 -0.691599 0.411855
4 N 1 Y -0.198359 0.337424 0.566607 -0.294182 -0.694125
5 N 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 C 2 S 0.000006 0.000023 -0.171322 -0.051660 -0.000008
7 C 2 S -0.000037 -0.000161 1.226239 0.376776 0.000061
8 C 2 X -0.565393 0.330922 0.000058 -0.000253 1.127178
9 C 2 Y -0.000029 -0.000083 -0.030473 0.950534 0.000250
10 C 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 N 3 S -0.118306 -0.021997 0.013233 0.027323 0.037880
12 N 3 S 0.899487 0.158448 -0.085819 -0.230690 -0.341559
13 N 3 X -0.003204 0.642604 0.459932 0.691406 0.412305
14 N 3 Y 0.198244 -0.337589 0.566554 -0.294731 0.693874
15 N 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
16 H 4 S 0.325490 0.809547 -0.014781 -0.508049 -0.118898
17 H 5 S -0.000052 0.000027 -0.618142 0.564520 0.000230
18 H 6 S -0.748953 0.232810 -0.504653 0.260983 -0.455364
19 H 7 S 0.748994 -0.232712 -0.504631 0.260627 0.455550
20 H 8 S -0.325703 -0.809527 -0.014987 -0.508211 0.118591
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6952440194
TWO ELECTRON ENERGY = 122.9489113878
NUCLEAR REPULSION ENERGY = 78.7058928763
------------------
TOTAL ENERGY = -149.0404397552
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9489113878
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3153113819
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7058928763
------------------
TOTAL POTENTIAL ENERGY = -296.6605071177
TOTAL KINETIC ENERGY = 147.6200673625
VIRIAL RATIO (V/T) = 2.0096218110
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -104.7974210974
BARE H ENERGY= -350.6952440194
ELECTRONIC ENERGY = -227.7463325584
KINETIC ENERGY= 147.6200673625
N-N REPULSION= 78.7058928763
TOTAL ENERGY= -149.0404396821
SIGMA PART(1+2)= -205.8544345303
(K,V1,2)= 140.8289390916 -450.5293474039 103.8459737821
PI PART(1+2)= -21.8918980281
(K,V1,2)= 6.7911282709 -47.7859639779 19.1029376789
SIGMA SKELETON, ERROR= -127.1485416540 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.725924 1.275789 -0.000578 0.626858 0.716231
2 -0.000668 -0.000824 2.001702 0.574298 0.287951
3 1.275697 0.726016 -0.000578 0.626927 0.716170
4 -0.000176 -0.000309 -0.000005 0.034040 0.075590
5 0.000000 0.000003 -0.000526 0.025581 0.000000
6 -0.000300 -0.000183 -0.000005 0.039128 0.064234
7 -0.000168 -0.000315 -0.000005 0.039121 0.064237
8 -0.000309 -0.000176 -0.000005 0.034046 0.075587
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.407577 0.490326 0.571200 0.597011 0.314100
2 0.620155 0.433506 0.400922 0.137054 0.492070
3 0.407566 0.490435 0.571246 0.596910 0.314055
4 0.051830 0.201513 -0.000039 0.290982 0.039422
5 0.223331 0.128976 0.000000 0.000000 0.431679
6 0.118848 0.026844 0.228364 0.043558 0.184621
7 0.118859 0.026839 0.228346 0.043551 0.184644
8 0.051834 0.201560 -0.000039 0.290933 0.039409
11 12
2.000000 2.000000
1 0.649066 1.000021
2 0.701826 0.000000
3 0.649109 0.999979
4 0.000000 0.000000
5 0.000000 0.000000
6 0.000000 0.000000
7 0.000000 0.000000
8 0.000000 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44490 1.22607
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.17286 1.16985
5 N 1 Z 1.64909 1.64328
6 C 2 S 1.99614 1.98933
7 C 2 S 1.09244 1.01766
8 C 2 X 0.82526 0.90520
9 C 2 Y 1.03233 1.05133
10 C 2 Z 0.70183 0.71344
11 N 3 S 1.99701 1.99305
12 N 3 S 1.44489 1.22606
13 N 3 X 1.10967 1.12800
14 N 3 Y 1.17287 1.16986
15 N 3 Z 1.64909 1.64328
16 H 4 S 0.69285 0.77749
17 H 5 S 0.80904 0.87314
18 H 6 S 0.70511 0.78721
19 H 7 S 0.70511 0.78721
20 H 8 S 0.69284 0.77748
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2508188
2 0.4697519 4.4404105
3 -0.0270992 0.4697547 6.2508150
4 0.3529402 -0.0305260 0.0015460 0.3975239
5 -0.0220403 0.3879558 -0.0220370 -0.0042956 0.4692147
6 -0.0027261 -0.0294159 0.3503342 -0.0000007 0.0022704
7 0.3503324 -0.0294135 -0.0027256 -0.0242240 0.0022705
8 0.0015460 -0.0305246 0.3529427 -0.0001157 -0.0042947
6 7 8
6 0.4076802
7 0.0011910 0.4076805
8 -0.0242239 -0.0000007 0.3975117
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.373524 -0.373524 7.160252 -0.160252
2 C 5.647993 0.352007 5.676954 0.323046
3 N 7.373531 -0.373531 7.160252 -0.160252
4 H 0.692848 0.307152 0.777486 0.222514
5 H 0.809044 0.190956 0.873144 0.126856
6 H 0.705109 0.294891 0.787215 0.212785
7 H 0.705111 0.294889 0.787215 0.212785
8 H 0.692841 0.307159 0.777482 0.222518
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 1.329 1.424 1 3 2.350 0.128 1 4 1.022 0.878
1 7 1.023 0.882 2 3 1.329 1.424 2 5 1.100 0.935
3 6 1.023 0.882 3 8 1.022 0.878
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 N 3.324 3.324 0.000
2 C 3.825 3.825 0.000
3 N 3.324 3.324 0.000
4 H 0.906 0.906 0.000
5 H 0.964 0.964 0.000
6 H 0.913 0.913 0.000
7 H 0.913 0.913 0.000
8 H 0.906 0.906 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
-0.000101 -0.005057 0.000000 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
-0.000053 -0.062068 0.000000 0.062068
...... END OF PROPERTY EVALUATION ......
Polymethin, 4 pi-Elektronen, S0,
CI
Input
!
! Polymethin, 4 pi-Elektronen, S0, CI
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=OPTIMIZE COORD=ZMTMPC
CITYP=GUGA $END
$SYSTEM TIMLIM=10 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$CIDRT GROUP=C1 IEXCIT=1 NFZC=3 NDOC=9
NALP=0 NVAL=8 $END
$GUGDIA NSTATE=3 $END
$GUGDM IROOT=1 NFLGDM(1)=1 $END
$GUGDM2 WSTATE(1)=1.0,0.0 $END
$GUESS GUESS=MOREAD NORB=20 $END
$DATA
Polymethin, 4 pi-Elektronen, S0, CI
C1
N 00000.0000 0 00000.0000 0 00000.0000 0 0 0 0
C 00001.3488 1 00000.0000 0 00000.0000 0 1 0 0
N 00001.3487 1 00122.5834 1 00000.0000 0 2 1 0
H 00000.9903 1 00119.4361 1 00179.9999 1 1 2 3
H 00001.1111 1 00118.7020 1 00180.0000 1 2 1 3
H 00000.9917 1 00122.3903 1 00000.0000 1 3 2 1
H 00000.9919 1 00122.3841 1 00179.9999 1 1 2 5
H 00000.9901 1 00119.3931 1 00000.0000 1 3 2 5
$END
--- OPTIMIZED RHF MO-S --- GENERATED AT 8:22:57 LT 10-DEC-2002
E= -149.0404397552, E(NUC)= 78.7058928763
$VEC
1 1-5.9986940E-01-1.2044646E-02-4.3652498E-04 4.7042868E-04 0.0000000E+00
1 2 6.3524551E-05-8.2065681E-04-4.0985608E-03-3.1218425E-04 0.0000000E+00
1 3 7.9520056E-01 1.6020677E-02-6.5769773E-04-5.3634987E-04 0.0000000E+00
1 4 2.3431186E-03 2.3525652E-05-3.0597802E-03 2.2977627E-03-3.0967358E-03
2 1 7.9518875E-01 1.6162460E-02 8.6607796E-04-3.0630897E-04 0.0000000E+00
bis
19 4-5.0804873E-01 5.6452039E-01 2.6098334E-01 2.6062698E-01-5.0821078E-01
20 1-3.7864677E-02 3.4142074E-01 4.1185487E-01-6.9412513E-01 0.0000000E+00
20 2-8.2707946E-06 6.1227441E-05 1.1271780E+00 2.5009109E-04 0.0000000E+00
20 3 3.7880397E-02-3.4155938E-01 4.1230517E-01 6.9387441E-01 0.0000000E+00
20 4-1.1889804E-01 2.2993329E-04-4.5536376E-01 4.5555000E-01 1.1859082E-01
$END
Output
-----------------------------
properties for the CI density
-----------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6952219044
TWO ELECTRON ENERGY = 122.9489021302
NUCLEAR REPULSION ENERGY = 78.7058800188
------------------
TOTAL ENERGY = -149.0404397555
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9489021302
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3152874768
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7058800188
------------------
TOTAL POTENTIAL ENERGY = -296.6605053278
TOTAL KINETIC ENERGY = 147.6200655723
VIRIAL RATIO (V/T) = 2.0096218233
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.597009 0.571199 0.716229 0.314100 0.626859
2 0.137052 0.400924 0.287951 0.492070 0.574298
3 0.596912 0.571246 0.716171 0.314054 0.626926
4 0.290981 -0.000039 0.075590 0.039422 0.034040
5 0.000000 0.000000 0.000000 0.431679 0.025581
6 0.043559 0.228364 0.064234 0.184621 0.039128
7 0.043552 0.228345 0.064237 0.184645 0.039121
8 0.290934 -0.000039 0.075587 0.039409 0.034046
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.407578 0.649067 1.264585 1.000020 0.737128
2 0.620155 0.701825 -0.000824 0.000000 -0.000668
3 0.407566 0.649108 0.737220 0.999980 1.264493
4 0.051831 0.000000 -0.000306 0.000000 -0.000178
5 0.223330 0.000000 0.000003 0.000000 0.000000
6 0.118848 0.000000 -0.000185 0.000000 -0.000298
7 0.118858 0.000000 -0.000313 0.000000 -0.000170
8 0.051834 0.000000 -0.000179 0.000000 -0.000306
11 12 13 14 15
2.000000 2.000000 0.000000 0.000000 0.000000
1 -0.000578 0.490327 0.000000 0.000000 0.000000
2 2.001702 0.433507 0.000000 0.000000 0.000000
3 -0.000578 0.490433 0.000000 0.000000 0.000000
4 -0.000005 0.201513 0.000000 0.000000 0.000000
5 -0.000526 0.128977 0.000000 0.000000 0.000000
6 -0.000005 0.026844 0.000000 0.000000 0.000000
7 -0.000005 0.026840 0.000000 0.000000 0.000000
8 -0.000005 0.201559 0.000000 0.000000 0.000000
16 17 18 19 20
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
WARNING! CI POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR CI FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY CI WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44490 1.22607
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.17286 1.16985
5 N 1 Z 1.64909 1.64328
6 C 2 S 1.99614 1.98933
7 C 2 S 1.09244 1.01767
8 C 2 X 0.82526 0.90520
9 C 2 Y 1.03233 1.05133
10 C 2 Z 0.70183 0.71344
11 N 3 S 1.99701 1.99305
12 N 3 S 1.44489 1.22606
13 N 3 X 1.10967 1.12800
14 N 3 Y 1.17287 1.16986
15 N 3 Z 1.64909 1.64328
16 H 4 S 0.69285 0.77749
17 H 5 S 0.80904 0.87314
18 H 6 S 0.70511 0.78722
19 H 7 S 0.70511 0.78721
20 H 8 S 0.69284 0.77748
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2508188
2 0.4697519 4.4404104
3 -0.0270992 0.4697546 6.2508152
4 0.3529402 -0.0305259 0.0015460 0.3975237
5 -0.0220403 0.3879555 -0.0220369 -0.0042955 0.4692148
6 -0.0027262 -0.0294160 0.3503343 -0.0000007 0.0022704
7 0.3503323 -0.0294135 -0.0027256 -0.0242240 0.0022705
8 0.0015460 -0.0305245 0.3529426 -0.0001157 -0.0042946
6 7 8
6 0.4076805
7 0.0011910 0.4076809
8 -0.0242239 -0.0000007 0.3975117
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.373524 -0.373524 7.160252 -0.160252
2 C 5.647992 0.352008 5.676954 0.323046
3 N 7.373531 -0.373531 7.160252 -0.160252
4 H 0.692848 0.307152 0.777485 0.222515
5 H 0.809044 0.190956 0.873144 0.126856
6 H 0.705109 0.294891 0.787215 0.212785
7 H 0.705111 0.294889 0.787215 0.212785
8 H 0.692841 0.307159 0.777482 0.222518
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
-0.000101 -0.005058 0.000000 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
-0.000055 -0.062067 0.000000 0.062067
...... END OF PROPERTY EVALUATION ......
CPU TIME: STEP = 0.00 , TOTAL = 269.9 SECONDS ( 4.5 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 269.9 SECONDS ( 4.5 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
......END OF NBO ANALYSIS......
CPU TIME: STEP = 0.00 , TOTAL = 269.9 SECONDS ( 4.5 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 269.9 SECONDS ( 4.5 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------------
properties for the Relaxed CI density
-------------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6952218797
TWO ELECTRON ENERGY = 122.9489021055
NUCLEAR REPULSION ENERGY = 78.7058800188
------------------
TOTAL ENERGY = -149.0404397555
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9489021055
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3152874156
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7058800188
------------------
TOTAL POTENTIAL ENERGY = -296.6605052913
TOTAL KINETIC ENERGY = 147.6200655359
VIRIAL RATIO (V/T) = 2.0096218235
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.597009 0.571199 0.716229 0.314100 0.626859
2 0.137052 0.400924 0.287951 0.492070 0.574298
3 0.596912 0.571246 0.716171 0.314054 0.626926
4 0.290981 -0.000039 0.075590 0.039422 0.034040
5 0.000000 0.000000 0.000000 0.431679 0.025581
6 0.043559 0.228364 0.064234 0.184621 0.039128
7 0.043552 0.228345 0.064237 0.184645 0.039121
8 0.290934 -0.000039 0.075587 0.039409 0.034046
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.407578 0.649067 1.264585 1.000020 0.737128
2 0.620155 0.701825 -0.000824 0.000000 -0.000668
3 0.407566 0.649108 0.737220 0.999980 1.264493
4 0.051831 0.000000 -0.000306 0.000000 -0.000178
5 0.223330 0.000000 0.000003 0.000000 0.000000
6 0.118848 0.000000 -0.000185 0.000000 -0.000298
7 0.118858 0.000000 -0.000313 0.000000 -0.000170
8 0.051834 0.000000 -0.000179 0.000000 -0.000306
11 12 13 14 15
2.000000 2.000000 0.000000 0.000000 0.000000
1 -0.000578 0.490327 0.000000 0.000000 0.000000
2 2.001702 0.433507 0.000000 0.000000 0.000000
3 -0.000578 0.490433 0.000000 0.000000 0.000000
4 -0.000005 0.201513 0.000000 0.000000 0.000000
5 -0.000526 0.128977 0.000000 0.000000 0.000000
6 -0.000005 0.026844 0.000000 0.000000 0.000000
7 -0.000005 0.026840 0.000000 0.000000 0.000000
8 -0.000005 0.201559 0.000000 0.000000 0.000000
16 17 18 19 20
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
WARNING! CI POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR CI FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY CI WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44490 1.22607
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.17286 1.16985
5 N 1 Z 1.64909 1.64328
6 C 2 S 1.99614 1.98933
7 C 2 S 1.09244 1.01767
8 C 2 X 0.82526 0.90520
9 C 2 Y 1.03233 1.05133
10 C 2 Z 0.70183 0.71344
11 N 3 S 1.99701 1.99305
12 N 3 S 1.44489 1.22606
13 N 3 X 1.10967 1.12800
14 N 3 Y 1.17287 1.16986
15 N 3 Z 1.64909 1.64328
16 H 4 S 0.69285 0.77749
17 H 5 S 0.80904 0.87314
18 H 6 S 0.70511 0.78722
19 H 7 S 0.70511 0.78721
20 H 8 S 0.69284 0.77748
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2508188
2 0.4697519 4.4404104
3 -0.0270992 0.4697546 6.2508151
4 0.3529402 -0.0305259 0.0015460 0.3975237
5 -0.0220403 0.3879555 -0.0220369 -0.0042955 0.4692148
6 -0.0027262 -0.0294160 0.3503343 -0.0000007 0.0022704
7 0.3503323 -0.0294135 -0.0027256 -0.0242240 0.0022705
8 0.0015460 -0.0305245 0.3529426 -0.0001157 -0.0042946
6 7 8
6 0.4076805
7 0.0011910 0.4076810
8 -0.0242239 -0.0000007 0.3975117
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.373524 -0.373524 7.160252 -0.160252
2 C 5.647993 0.352007 5.676954 0.323046
3 N 7.373531 -0.373531 7.160252 -0.160252
4 H 0.692848 0.307152 0.777485 0.222515
5 H 0.809044 0.190956 0.873144 0.126856
6 H 0.705109 0.294891 0.787215 0.212785
7 H 0.705111 0.294889 0.787215 0.212785
8 H 0.692841 0.307159 0.777482 0.222518
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
-0.000101 -0.005058 0.000000 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
-0.000055 -0.062067 0.000000 0.062067
...... END OF PROPERTY EVALUATION ......
Die Geometrie des Moleküls müsste eigentlich C2v
entsprechen. In der
folgenden Rechnung wird C2v vorausgesetzt (Achtung, Nummerierung
ändert sich!).
Mit C2v:

Input
!
! Polymethin, 4 pi-Elektronen
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=OPTIMIZE
COORD=ZMT $END
$SYSTEM TIMLIM=10 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Polymethin, 4 pi-Elektronen
CNV 2
N
C 1 1.33
N 2 1.33 1 120.00
H 1 1.02 2 120.00 3 180.00
H 2 1.10 1 120.00 3 180.00
H 3 1.02 2 120.00 1 0.00
H 1 1.02 2 120.00 5 180.00
H 3 1.02 2 120.00 5 0.00
$END
Output
NSERCH= 10 ENERGY= -149.0404398
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX
DE/DY DE/DZ
--------------------------------------------------------------
1 N 7.0 0.0000499 0.0000000
0.0000061
2 N 7.0 -0.0000499 0.0000000
0.0000061
3 C 6.0 0.0000000 0.0000000
-0.0000194
4 H 1.0 -0.0000755 0.0000000
0.0000363
5 H 1.0 0.0000755 0.0000000
0.0000363
6 H 1.0 0.0000000 0.0000000
-0.0000120
7 H 1.0 -0.0000054 0.0000000
-0.0000267
8 H 1.0 0.0000054 0.0000000
-0.0000267
MAXIMUM GRADIENT = 0.0000755 RMS
GRADIENT = 0.0000296
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Polymethin, 4 pi-Elektronen
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N 7.0 -1.1750878737 0.0000000000
0.1905577406
C 6.0 0.0000000000
0.0000000000 -0.4307694266
H 1.0 -2.0426095716 0.0000000000
-0.3502741433
H 1.0 0.0000000000
0.0000000000 -1.5307303754
H 1.0 1.2521999445
0.0000000000 1.2107601887
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X
Y
Z
------------------------------------------------------------
N 7.0 1.1750878737
0.0000000000 0.1905577406
N 7.0 -1.1750878737 0.0000000000
0.1905577406
C 6.0 0.0000000000
0.0000000000 -0.4307694266
H 1.0 2.0426095716
0.0000000000 -0.3502741433
H 1.0 -2.0426095716 0.0000000000
-0.3502741433
H 1.0 0.0000000000
0.0000000000 -1.5307303754
H 1.0 -1.2521999445 0.0000000000
1.2107601887
H 1.0 1.2521999445
0.0000000000 1.2107601887
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
N
C 1 1.3292400
N 2 1.3292400 1 124.2647472
H 1 1.0222979 2 120.1920633 3 180.0000000 0
H 2 1.0999609 1 117.8676264 3 180.0000000 0
H 3 1.0231126 2 122.1901127 1 0.0000000 0
H 1 1.0231126 2 122.1901127 5 180.0000000 0
H 3 1.0222979 2 120.1920633 5 0.0000000 0
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
N
N C
H
1 N 0.0000000 2.3501757 * 1.3292400 *
1.0222979 *
2 N 2.3501757 * 0.0000000 1.3292400 *
3.2628325
3 C 1.3292400 * 1.3292400 * 0.0000000
2.0441950 *
4 H 1.0222979 * 3.2628325 2.0441950 *
0.0000000
5 H 3.2628325 1.0222979 * 2.0441950 *
4.0852191
6 H 2.0841459 * 2.0841459 * 1.0999609 *
2.3591801 *
7 H 2.6329716 * 1.0231126 * 2.0646124 *
3.6459015
8 H 1.0231126 * 2.6329716 * 2.0646124 *
1.7497359 *
H
H H
H
1 N 3.2628325 2.0841459 * 2.6329716 *
1.0231126 *
2 N 1.0222979 * 2.0841459 * 1.0231126 *
2.6329716 *
3 C 2.0441950 * 1.0999609 * 2.0646124 *
2.0646124 *
4 H 4.0852191 2.3591801 * 3.6459015
1.7497359 *
5 H 0.0000000 2.3591801 * 1.7497359 *
3.6459015
6 H 2.3591801 * 0.0000000 3.0139302
3.0139302
7 H 1.7497359 * 3.0139302 0.0000000
2.5043999 *
8 H 3.6459015 3.0139302
2.5043999 * 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 78.7053044253
ELECTRONIC ENERGY = -227.7457441827
TOTAL ENERGY = -149.0404397574
vorher (C1): -149.0404397555
------------------
MOLECULAR ORBITALS
------------------
1 2
3 4
5
-15.8827
-15.8826 -11.6415 -1.5073 -1.3961
B1 A1
A1 A1
B1
1 N 1 S 0.704339 0.704325
0.000074 -0.134124 -0.155271
2 N 1 S 0.014170 0.014336 -0.003643
0.454176 0.545080
3 N 1 X -0.000552 -0.000797 0.002081
-0.078854 -0.014517
4 N 1 Y 0.000000 0.000000
0.000000 0.000000 0.000000
5 N 1 Z -0.000508 -0.000238 0.000934
-0.033112 -0.021739
6 N 2 S -0.704339 0.704325 0.000074
-0.134124 0.155271
7 N 2 S -0.014170 0.014336 -0.003643
0.454176 -0.545080
8 N 2 X -0.000552 0.000797 -0.002081
0.078854 -0.014517
9 N 2 Y 0.000000 0.000000
0.000000 0.000000 0.000000
10 N 2 Z 0.000508 -0.000238 0.000934
-0.033112 0.021739
11 C 3 S 0.000000 0.000458 0.995644
-0.142188 0.000000
12 C 3 S 0.000000 -0.005918 0.020712
0.354629 0.000000
13 C 3 X -0.004138 0.000000 0.000000
0.000000 0.220235
14 C 3 Y 0.000000 0.000000 0.000000
0.000000 0.000000
15 C 3 Z 0.000000 -0.002253 0.000855
0.081286 0.000000
16 H 4 S -0.002747 -0.002717 -0.000308
0.055030 0.095530
17 H 5 S 0.002747 -0.002717 -0.000308
0.055030 -0.095530
18 H 6 S 0.000000 0.000170 -0.003898
0.046250 0.000000
19 H 7 S 0.002705 -0.002748 -0.000308
0.058234 -0.091173
20 H 8 S -0.002705 -0.002748 -0.000308
0.058234 0.091173
6 7
8 9
10
-1.0936
-0.9847 -0.9401 -0.8635
-0.8121
A1 A1
B1 B1
A1
1 N 1 S 0.065421 0.014938 -0.013131
0.007169 -0.014816
2 N 1 S -0.266899 -0.072535 0.075599
-0.023922 0.064040
3 N 1 X -0.214878 -0.322892 0.191154
0.424468 -0.046409
4 N 1 Y 0.000000 0.000000
0.000000 0.000000 0.000000
5 N 1 Z -0.179865 0.216839 0.391719
-0.218246 -0.359755
6 N 2 S 0.065421 0.014938
0.013131 -0.007169 -0.014816
7 N 2 S -0.266899 -0.072535 -0.075599
0.023922 0.064040
8 N 2 X 0.214878 0.322892
0.191154 0.424468 0.046409
9 N 2 Y 0.000000 0.000000
0.000000 0.000000 0.000000
10 N 2 Z -0.179865 0.216839 -0.391719
0.218246 -0.359755
11 C 3 S -0.157243 -0.013484 0.000000
0.000000 0.019074
12 C 3 S 0.513018 0.035983 0.000000
0.000000 -0.090499
13 C 3 X 0.000000 0.000000 -0.376535
-0.247384 0.000000
14 C 3 Y 0.000000 0.000000 0.000000
0.000000 0.000000
15 C 3 Z -0.134499 0.356349 0.000000
0.000000 0.436403
16 H 4 S -0.111929 -0.235821 -0.003877
0.317475 0.132830
17 H 5 S -0.111929 -0.235821 0.003877
-0.317475 0.132830
18 H 6 S 0.219858 -0.176689 0.000000
0.000000 -0.391014
19 H 7 S -0.164871 0.088634 -0.265543
0.131714 -0.263091
20 H 8 S -0.164871 0.088634 0.265543
-0.131714 -0.263091
11 12=HOMO
13=LUMO 14
15
-0.7821
-0.6124
-0.0098 0.2702
0.3040
B2 A2
B2 A1
A1
1 N 1 S 0.000000 0.000000
0.000000 -0.098295 -0.085933
2 N 1 S 0.000000 0.000000
0.000000 0.725027 0.618253
3 N 1 X 0.000000 0.000000
0.000000 0.113620 -0.045143
4 N 1 Y 0.517759 0.712257 -0.518997
0.000000 0.000000
5 N 1 Z 0.000000 0.000000
0.000000 -0.264464 0.289834
6 N 2 S 0.000000 0.000000
0.000000 -0.098295 -0.085933
7 N 2 S 0.000000 0.000000
0.000000 0.725027 0.618253
8 N 2 X 0.000000 0.000000
0.000000 -0.113620 0.045143
9 N 2 Y 0.517759 -0.712257 -0.518997
0.000000 0.000000
10 N 2 Z 0.000000 0.000000 0.000000
-0.264464 0.289834
11 C 3 S 0.000000 0.000000 0.000000
0.033519 0.119850
12 C 3 S 0.000000 0.000000 0.000000
-0.185422 -0.776887
13 C 3 X 0.000000 0.000000 0.000000
0.000000 0.000000
14 C 3 Y 0.495657 0.000000 0.919063
0.000000 0.000000
15 C 3 Z 0.000000 0.000000 0.000000
-0.637412 0.330635
16 H 4 S 0.000000 0.000000 0.000000
-0.722383 -0.127761
17 H 5 S 0.000000 0.000000 0.000000
-0.722383 -0.127761
18 H 6 S 0.000000 0.000000 0.000000
-0.486225 0.821810
19 H 7 S 0.000000 0.000000 0.000000
-0.054702 -0.646633
20 H 8 S 0.000000 0.000000 0.000000
-0.054702 -0.646633
HOMO-1
HOMO
LUMO
16 17
18 19
20
0.3169 0.4156 0.4330
0.6132 0.6313
B1 B1
A1 A1
B1
1 N 1 S -0.118288 0.022058 0.013179
0.027316 0.037857
2 N 1 S 0.899379 -0.158949 -0.085397
-0.230627 -0.341346
3 N 1 X -0.003568 -0.642714 0.459797
0.691543 0.412097
4 N 1 Y 0.000000 0.000000
0.000000 0.000000 0.000000
5 N 1 Z 0.198413 0.337406
0.566677 -0.294437 0.694016
6 N 2 S 0.118288 -0.022058 0.013179
0.027316 -0.037857
7 N 2 S -0.899379 0.158949 -0.085397
-0.230627 0.341346
8 N 2 X -0.003568 -0.642714 -0.459797
-0.691543 0.412097
9 N 2 Y 0.000000 0.000000
0.000000 0.000000 0.000000
10 N 2 Z -0.198413 -0.337406 0.566677
-0.294437 -0.694016
11 C 3 S 0.000000 0.000000 -0.171259
-0.051661 0.000000
12 C 3 S 0.000000 0.000000 1.225795
0.376785 0.000000
13 C 3 X -0.565697 -0.330591 0.000000
0.000000 1.127092
14 C 3 Y 0.000000 0.000000 0.000000
0.000000 0.000000
15 C 3 Z 0.000000 0.000000 -0.030496
0.950451 0.000000
16 H 4 S -0.325191 0.809760 -0.015050
-0.508263 0.118694
17 H 5 S 0.325191 -0.809760 -0.015050
-0.508263 -0.118694
18 H 6 S 0.000000 0.000000 -0.617890
0.564437 0.000000
19 H 7 S 0.749046 0.232356 -0.504982
0.260738 0.455565
20 H 8 S -0.749046 -0.232356 -0.504982
0.260738 -0.455565
------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6939552163
TWO ELECTRON ENERGY = 122.9482110336
NUCLEAR REPULSION ENERGY = 78.7053044253
------------------
TOTAL ENERGY = -149.0404397574
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9482110336
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3140197559
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7053044253
------------------
TOTAL POTENTIAL ENERGY = -296.6605042970
TOTAL KINETIC ENERGY = 147.6200645396
VIRIAL RATIO (V/T) = 2.0096218304
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -104.7975330232
BARE H ENERGY= -350.6939552163
ELECTRONIC ENERGY = -227.7457441198
KINETIC ENERGY= 147.6200645396
N-N REPULSION= 78.7053044253
TOTAL ENERGY= -149.0404396945
SIGMA PART(1+2)= -205.8539578269
(K,V1,2)= 140.8289137623 -450.5282509715 103.8453793823
PI PART(1+2)= -21.8917862929
(K,V1,2)= 6.7911507772 -47.7857687844 19.1028317143
SIGMA SKELETON, ERROR= -127.1486534016 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2 3
4 5
2.000000 2.000000
2.000000 2.000000 2.000000
1 1.000809 1.000904 -0.000578
0.626896 0.716203
2 1.000809 1.000904 -0.000578
0.626896 0.716203
3 -0.000665 -0.000827 2.001701
0.574270 0.287904
4 -0.000242 -0.000243 -0.000005
0.034053 0.075603
5 -0.000242 -0.000243 -0.000005
0.034053 0.075603
6 0.000000 0.000003 -0.000526
0.025578 0.000000
7 -0.000234 -0.000249 -0.000005
0.039127 0.064242
8 -0.000234 -0.000249 -0.000005
0.039127 0.064242
6
7 8
9 10
2.000000 2.000000
2.000000 2.000000 2.000000
1 0.407622 0.490327 0.571215
0.596973 0.314081
2 0.407622 0.490327 0.571215
0.596973 0.314081
3 0.620103 0.433600 0.401024
0.136985 0.492072
4 0.051870 0.201486 -0.000039
0.290942 0.039421
5 0.051870 0.201486 -0.000039
0.290942 0.039421
6 0.223245 0.129052 0.000000
0.000000 0.431680
7 0.118834 0.026860 0.228313
0.043592 0.184622
8 0.118834 0.026860 0.228313
0.043592 0.184622
11
12
2.000000 2.000000
1 0.649099 1.000000
2 0.649099 1.000000
3 0.701802 0.000000
4 0.000000 0.000000
5 0.000000 0.000000
6 0.000000 0.000000
7 0.000000 0.000000
8 0.000000 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44488 1.22604
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.64910 1.64329
5 N 1 Z 1.17289 1.16988
6 N 2 S 1.99701 1.99305
7 N 2 S 1.44488 1.22604
8 N 2 X 1.10967 1.12800
9 N 2 Y 1.64910 1.64329
10 N 2 Z 1.17289 1.16988
11 C 3 S 1.99614 1.98933
12 C 3 S 1.09246 1.01768
13 C 3 X 0.82525 0.90518
14 C 3 Y 0.70180 0.71342
15 C 3 Z 1.03232 1.05132
16 H 4 S 0.69285 0.77750
17 H 5 S 0.69285 0.77750
18 H 6 S 0.80903 0.87313
19 H 7 S 0.70510 0.78721
20 H 8 S 0.70510 0.78721
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1
2 3
4 5
1 6.2508258
2 -0.0270972 6.2508258
3 0.4697531 0.4697531 4.4403752
4 0.3529497 0.0015459 -0.0305225
0.3975164
5 0.0015459 0.3529497 -0.0305225
-0.0001157 0.3975164
6 -0.0220366 -0.0220366 0.3879473
-0.0042942 -0.0042942
7 -0.0027249 0.3503362 -0.0294076
-0.0000007 -0.0242332
8 0.3503362 -0.0027249 -0.0294076
-0.0242332 -0.0000007
6
7 8
6 0.4692059
7 0.0022701 0.4076721
8 0.0022701 0.0011903 0.4076721
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 N 7.373552
-0.373552 7.160263
-0.160263
2 N 7.373552
-0.373552 7.160263
´-0.160263
3 C 5.647968
0.352032
5.676924 0.323076
4 H 0.692846 0.307154
0.777496 0.222504
5 H 0.692846 0.307154
0.777496 0.222504
6 H 0.809032 0.190968
0.873131 0.126869
7 H 0.705102 0.294898
0.787213 0.212787
8 H 0.705102 0.294898
0.787213 0.212787
Man sieht, dass die Ladung alterniert und zahlenmäßig
nahezu gleich ist (obwohl C und N).
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 2 2.350 0.128
1 3
1.329 1.424 1 4 1.022 0.878
1 8 1.023 0.882
2 3
1.329 1.424 2 5 1.022 0.878
2 7 1.023 0.882 3 6
1.100 0.935
Bindungsausgleich!
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 N 3.324 3.324 0.000
2 N 3.324 3.324 0.000
3 C 3.825 3.825 0.000
4 H 0.906 0.906 0.000
5 H 0.906 0.906 0.000
6 H 0.964 0.964 0.000
7 H 0.913 0.913 0.000
8 H 0.913 0.913 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X
Y Z (BOHR) CHARGE
0.000000 0.000000
0.015073 1.00 (A.U.)
DX
DY DZ
/D/ (DEBYE)
0.000000 0.000000
-0.062001 0.062001
Dipolmoment entlang der C2-Drehachse.
...... END OF PROPERTY EVALUATION ......
Mit CI:
THE 6 LOWEST DIAGONAL ELEMENTS OF THE HAMILTONIAN ARE
-149.0404398 (CSF 1) -148.5027183 (CSF 2) -148.2324892 (CSF 24)
-148.1843781 (CSF 20) -148.1542197 (CSF 9) -148.0849819 (CSF 16)
RESTARTING WITH PREVIOUS CI EIGENVECTORS...
STATE # 1 ENERGY = -149.040439757
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
1 1.000000 22222222200000000
STATE # 2 ENERGY = -148.558547684
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
2 0.963961 22222221210000000
3 -0.067333 22222122200000001
4 0.119809 22221222200000001
6 0.072040 22222122200001000
7 -0.051826 22221222200001000
10 -0.071828 22221222200010000
12 -0.057488 22222212200000010
13 0.081917 22212222200000010
18 -0.085415 22122222200000100
24 0.080821 22222212201000000
STATE # 3 ENERGY = -148.259025479
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
2 -0.058049 22222221210000000
6 0.056138 22222122200001000
7 0.158322 22221222200001000
9 -0.309886 22222122200010000
10 -0.155734 22221222200010000
20 0.196275 22222212200100000
21 0.119711 22212222200100000
24 0.880578 22222212201000000
25 0.095017 22212222201000000
...... END OF CI-MATRIX DIAGONALIZATION ......
MAXIMUM GRADIENT = 0.0000755 RMS GRADIENT = 0.0000296
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
Polymethin, 4 pi-Elektronen, S0, C2v, CI
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
N 7.0 -1.1750877595 0.0000000000 0.1905576725
C 6.0 0.0000000000 0.0000000000 -0.4307692971
H 1.0 -2.0426094297 0.0000000000 -0.3502743009
H 1.0 0.0000000000 0.0000000000 -1.5307300432
H 1.0 1.2522004211 0.0000000000 1.2107601834
THE CURRENT FULLY SUBSTITUTED Z-MATRIX IS
N
C 1 1.3292398
N 2 1.3292398 1 124.2647577
H 1 1.0222979 2 120.1920634 3 180.0000000 0
H 2 1.0999607 1 117.8676212 3 180.0000000 0
H 3 1.0231127 2 122.1901402 1 0.0000000 0
H 1 1.0231127 2 122.1901402 5 180.0000000 0
H 3 1.0222979 2 120.1920634 5 0.0000000 0
-----------------------------
properties for the CI density
-----------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6939664695
TWO ELECTRON ENERGY = 122.9482162983
NUCLEAR REPULSION ENERGY = 78.7053104138
------------------
TOTAL ENERGY = -149.0404397574
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9482162983
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3140312103
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7053104138
------------------
TOTAL POTENTIAL ENERGY = -296.6605044982
TOTAL KINETIC ENERGY = 147.6200647408
VIRIAL RATIO (V/T) = 2.0096218290
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.626896 0.407623 1.000000 0.596974 0.716203
2 0.626896 0.407623 1.000000 0.596974 0.716203
3 0.574270 0.620102 0.000000 0.136984 0.287904
4 0.034053 0.051870 0.000000 0.290942 0.075603
5 0.034053 0.051870 0.000000 0.290942 0.075603
6 0.025578 0.223245 0.000000 0.000000 0.000000
7 0.039127 0.118834 0.000000 0.043592 0.064242
8 0.039127 0.118834 0.000000 0.043592 0.064242
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.490327 0.314081 0.571215 0.649098 1.000809
2 0.490327 0.314081 0.571215 0.649098 1.000809
3 0.433600 0.492072 0.401023 0.701804 -0.000665
4 0.201486 0.039421 -0.000039 0.000000 -0.000242
5 0.201486 0.039421 -0.000039 0.000000 -0.000242
6 0.129053 0.431680 0.000000 0.000000 0.000000
7 0.026860 0.184622 0.228313 0.000000 -0.000234
8 0.026860 0.184622 0.228313 0.000000 -0.000234
11 12 13 14 15
2.000000 2.000000 0.000000 0.000000 0.000000
1 1.000904 -0.000578 0.000000 0.000000 0.000000
2 1.000904 -0.000578 0.000000 0.000000 0.000000
3 -0.000827 2.001701 0.000000 0.000000 0.000000
4 -0.000243 -0.000005 0.000000 0.000000 0.000000
5 -0.000243 -0.000005 0.000000 0.000000 0.000000
6 0.000003 -0.000526 0.000000 0.000000 0.000000
7 -0.000249 -0.000005 0.000000 0.000000 0.000000
8 -0.000249 -0.000005 0.000000 0.000000 0.000000
16 17 18 19 20
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
WARNING! CI POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR CI FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY CI WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44488 1.22604
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.64910 1.64329
5 N 1 Z 1.17289 1.16988
6 N 2 S 1.99701 1.99305
7 N 2 S 1.44488 1.22604
8 N 2 X 1.10967 1.12800
9 N 2 Y 1.64910 1.64329
10 N 2 Z 1.17289 1.16988
11 C 3 S 1.99614 1.98933
12 C 3 S 1.09246 1.01768
13 C 3 X 0.82525 0.90518
14 C 3 Y 0.70180 0.71342
15 C 3 Z 1.03232 1.05132
16 H 4 S 0.69285 0.77750
17 H 5 S 0.69285 0.77750
18 H 6 S 0.80903 0.87313
19 H 7 S 0.70510 0.78721
20 H 8 S 0.70510 0.78721
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2508258
2 -0.0270973 6.2508258
3 0.4697532 0.4697532 4.4403747
4 0.3529496 0.0015459 -0.0305225 0.3975164
5 0.0015459 0.3529496 -0.0305225 -0.0001157 0.3975164
6 -0.0220366 -0.0220366 0.3879475 -0.0042942 -0.0042942
7 -0.0027249 0.3503361 -0.0294076 -0.0000007 -0.0242332
8 0.3503361 -0.0027249 -0.0294076 -0.0242332 -0.0000007
6 7 8
6 0.4692059
7 0.0022701 0.4076721
8 0.0022701 0.0011903 0.4076721
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.373552 -0.373552 7.160263 -0.160263
2 N 7.373552 -0.373552 7.160263 -0.160263
3 C 5.647968 0.352032 5.676925 0.323075
4 H 0.692846 0.307154 0.777496 0.222504
5 H 0.692846 0.307154 0.777496 0.222504
6 H 0.809032 0.190968 0.873131 0.126869
7 H 0.705102 0.294898 0.787213 0.212787
8 H 0.705102 0.294898 0.787213 0.212787
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.015073 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.061998 0.061998
...... END OF PROPERTY EVALUATION ......
CPU TIME: STEP = 0.00 , TOTAL = 90.7 SECONDS ( 1.5 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 90.7 SECONDS ( 1.5 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
......END OF NBO ANALYSIS......
CPU TIME: STEP = 0.00 , TOTAL = 90.7 SECONDS ( 1.5 MIN)
WALL CLOCK TIME: STEP = 0.00 , TOTAL = 90.7 SECONDS ( 1.5 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
-------------------------------------
properties for the Relaxed CI density
-------------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -350.6939664807
TWO ELECTRON ENERGY = 122.9482163094
NUCLEAR REPULSION ENERGY = 78.7053104138
------------------
TOTAL ENERGY = -149.0404397574
ELECTRON-ELECTRON POTENTIAL ENERGY = 122.9482163094
NUCLEUS-ELECTRON POTENTIAL ENERGY = -498.3140312363
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 78.7053104138
------------------
TOTAL POTENTIAL ENERGY = -296.6605045130
TOTAL KINETIC ENERGY = 147.6200647556
VIRIAL RATIO (V/T) = 2.0096218289
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.626896 0.407623 1.000000 0.596974 0.716203
2 0.626896 0.407623 1.000000 0.596974 0.716203
3 0.574270 0.620102 0.000000 0.136984 0.287904
4 0.034053 0.051870 0.000000 0.290942 0.075603
5 0.034053 0.051870 0.000000 0.290942 0.075603
6 0.025578 0.223245 0.000000 0.000000 0.000000
7 0.039127 0.118834 0.000000 0.043592 0.064242
8 0.039127 0.118834 0.000000 0.043592 0.064242
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.490327 0.314081 0.571215 0.649098 1.000809
2 0.490327 0.314081 0.571215 0.649098 1.000809
3 0.433600 0.492072 0.401023 0.701804 -0.000665
4 0.201486 0.039421 -0.000039 0.000000 -0.000242
5 0.201486 0.039421 -0.000039 0.000000 -0.000242
6 0.129053 0.431680 0.000000 0.000000 0.000000
7 0.026860 0.184622 0.228313 0.000000 -0.000234
8 0.026860 0.184622 0.228313 0.000000 -0.000234
11 12 13 14 15
2.000000 2.000000 0.000000 0.000000 0.000000
1 1.000904 -0.000578 0.000000 0.000000 0.000000
2 1.000904 -0.000578 0.000000 0.000000 0.000000
3 -0.000827 2.001701 0.000000 0.000000 0.000000
4 -0.000243 -0.000005 0.000000 0.000000 0.000000
5 -0.000243 -0.000005 0.000000 0.000000 0.000000
6 0.000003 -0.000526 0.000000 0.000000 0.000000
7 -0.000249 -0.000005 0.000000 0.000000 0.000000
8 -0.000249 -0.000005 0.000000 0.000000 0.000000
16 17 18 19 20
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
WARNING! CI POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR CI FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY CI WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99701 1.99305
2 N 1 S 1.44488 1.22604
3 N 1 X 1.10967 1.12800
4 N 1 Y 1.64910 1.64329
5 N 1 Z 1.17289 1.16988
6 N 2 S 1.99701 1.99305
7 N 2 S 1.44488 1.22604
8 N 2 X 1.10967 1.12800
9 N 2 Y 1.64910 1.64329
10 N 2 Z 1.17289 1.16988
11 C 3 S 1.99614 1.98933
12 C 3 S 1.09246 1.01768
13 C 3 X 0.82525 0.90518
14 C 3 Y 0.70180 0.71342
15 C 3 Z 1.03232 1.05132
16 H 4 S 0.69285 0.77750
17 H 5 S 0.69285 0.77750
18 H 6 S 0.80903 0.87313
19 H 7 S 0.70510 0.78721
20 H 8 S 0.70510 0.78721
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2508258
2 -0.0270973 6.2508258
3 0.4697532 0.4697532 4.4403747
4 0.3529496 0.0015459 -0.0305225 0.3975164
5 0.0015459 0.3529496 -0.0305225 -0.0001157 0.3975164
6 -0.0220366 -0.0220366 0.3879475 -0.0042942 -0.0042942
7 -0.0027249 0.3503361 -0.0294076 -0.0000007 -0.0242332
8 0.3503361 -0.0027249 -0.0294076 -0.0242332 -0.0000007
6 7 8
6 0.4692059
7 0.0022701 0.4076721
8 0.0022701 0.0011903 0.4076721
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.373552 -0.373552 7.160263 -0.160263
2 N 7.373552 -0.373552 7.160263 -0.160263
3 C 5.647968 0.352032 5.676925 0.323075
4 H 0.692846 0.307154 0.777496 0.222504
5 H 0.692846 0.307154 0.777496 0.222504
6 H 0.809032 0.190968 0.873131 0.126869
7 H 0.705102 0.294898 0.787213 0.212787
8 H 0.705102 0.294898 0.787213 0.212787
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.015073 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.061998 0.061998
...... END OF PROPERTY EVALUATION ......
Input
!
! Polymethin, 4 pi-Elektronen, C2v, S0, CI
!
$CONTRL SCFTYP=NONE MULT=1 ICHARG=1 RUNTYP=TRANSITN COORD=UNIQUE CITYP=GUGA
$END
$SYSTEM TIMLIM=10 MEMORY=4000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$CIDRT1 GROUP=C2V IEXCIT=1 NFZC=3 NDOC=9 NALP=0 NVAL=8 $END
$TRANST IROOTS(1)=1,2 $END
$DATA
Polymethin, 4 pi-Elektronen, S0, C2v, CI
CNV 2
N 7.0 -1.1750877595 0.0000000000 0.1905576725
C 6.0 0.0000000000 0.0000000000 -0.4307692971
H 1.0 -2.0426094297 0.0000000000 -0.3502743009
H 1.0 0.0000000000 0.0000000000 -1.5307300432
H 1.0 1.2522004211 0.0000000000 1.2107601834
$END
--- OPTIMIZED RHF MO-S --- GENERATED AT 9:30:31 LT 11-DEC-2002
E= -149.0404397574, E(NUC)= 78.7053104138
$VEC1
1 1 7.04339380E-01 1.41697791E-02-5.51780448E-04 0.00000000E+00-5.08166241E-04
1 2-7.04339380E-01-1.41697791E-02-5.51780448E-04 0.00000000E+00 5.08166241E-04
bis
20 3 0.00000000E+00 0.00000000E+00 1.12709286E+00 0.00000000E+00 0.00000000E+00
20 4 1.18693337E-01-1.18693337E-01 0.00000000E+00 4.55564756E-01-4.55564756E-01
$END
Output
---------------------------
RADIATIVE TRANSITION MOMENT
---------------------------
NFZC = 20
NUMVEC= 1
NUMCI = 1
NOCC = 20
SELECT= NORMAL
IROOTS= 1 2
THE 4 LOWEST DIAGONAL ELEMENTS OF THE HAMILTONIAN ARE
-149.0404398 (CSF 1) -148.5027183 (CSF 2) -148.2324892 (CSF 24)
-148.1843781 (CSF 20)
SOLUTION FOUND WITH DIRECT METHOD
STATE # 1 ENERGY = -149.040439757
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
1 1.000000 22222222200000000
STATE # 2 ENERGY = -148.558547686
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
2 0.963961 22222221210000000
3 -0.067333 22222122200000001
4 0.119809 22221222200000001
6 0.072040 22222122200001000
7 -0.051826 22221222200001000
10 -0.071828 22221222200010000
12 -0.057488 22222212200000010
13 0.081917 22212222200000010
18 -0.085415 22122222200000100
24 0.080821 22222212201000000
...... END OF CI-MATRIX DIAGONALIZATION ......
---------------------
STORE CSF INFORMATION
--------------------
READING THE DRT FILE
TITLE=CIDRT1 13:02:58 LT 11-DEC-2002
NUMBER OF CONFIGURATIONS = 27
READING THE CI VECTOR FILE
RUN TITLE=Polymethin, 4 pi-Elektronen, S0, C2v, CI
DRT TITLE=CIDRT1 13:02:58 LT 11-DEC-2002
2 STATES WERE COMPUTED, NWKS= 27
STORING CSF AND CI COEF. INFORMATION FOR 2 STATES ON FILE 17.
ENERGIES ARE ...
-149.0404398 -148.5585477
...... DONE STORING CSF INFORMATION ......
------------------
TRANSITION MOMENTS
------------------
3883 WORDS OF MEMORY ARE REQUIRED.
RECOVER CI INFORMATION OF STATE 1. IROOTS= 1
RECOVER CI INFORMATION OF STATE 2. IROOTS= 2
---- LENGTH FORM ----
CI STATE NUMBER= 1 2 : STATE MULTIPLICITY= 1 1 : NUMBER OF CSF-S= 27 27
STATE ENERGIES -149.0404397574 -148.5585476857
TRANSITION ENERGY= 3.1707E+15 [1/SEC] = 105760.66 [1/CM] = 13.11 [EV]
X Y Z NORM
CENTER OF MASS = 0.000000 0.000000 0.000000 BOHR
TRANSITION DIPOLE = 0.000000 0.000000 0.659515 0.659515 E*BOHR
TRANSITION DIPOLE = 0.000000 0.000000 1.676332 1.676332 DEBYE
OSCILLATOR STRENGTH = 0.139736
EINSTEIN COEFFICIENTS: A= 1.0425E+09 1/SEC; B= 1.7652E+08 SEC/G
---- VELOCITY FORM ----
CI STATE NUMBER= 1 2 : STATE MULTIPLICITY= 1 1 : NUMBER OF CSF-S= 27 27
STATE ENERGIES -149.0404397574 -148.5585476857
TRANSITION ENERGY= 3.1707E+15 [1/SEC] = 105760.66 [1/CM] = 13.11 [EV]
= 94.6 nm
X Y Z NORM
DIPOLE VELOCITY <D/DQ> = 0.000000 0.000000 0.077255 0.077255 E/BOHR
OSCILLATOR STRENGTH IS 0.008257
CPU TIME: STEP = 0.05 , TOTAL = 1.0 SECONDS ( 0.0 MIN)
WALL CLOCK TIME: STEP = 0.05 , TOTAL = 1.0 SECONDS ( 0.0 MIN)
CPU UTILIZATION: STEP = 100.00%, TOTAL = 100.00%
6 p-Elektronen
Input, STO-6G, C2v
!
! Polymethin, 6 pi-Elektronen
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=1 RUNTYP=OPTIMIZE COORD=UNIQUE $END
$SYSTEM TIMLIM=100 MEMORY=40000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
Polymethin, 6 pi-Elektronen
CNV 2
N 7.0 -2.4117264674 0.0000000000 0.1986778687
C 6.0 -1.1993095172 0.0000000000 -0.3619209910
C 6.0 0.0000000000 0.0000000000 0.3389311049
H 1.0 -3.2584613107 0.0000000000 -0.3699670009
H 1.0 -1.1971026630 0.0000000000 -1.4550133177
H 1.0 0.0000000000 0.0000000000 1.4129813150
H 1.0 2.5276264541 0.0000000000 1.2125022265
$END
Output
16 17 18 19 20
-0.7041 -0.6996 -0.6546 -0.4681 -0.0304
B2 A1 A2 B2 A2
1 N 1 S 0.000000 0.011050 0.000000 0.000000 0.000000
2 N 1 S 0.000000 -0.055893 0.000000 0.000000 0.000000
3 N 1 X 0.000000 0.033015 0.000000 0.000000 0.000000
4 N 1 Y 0.437293 0.000000 0.582661 -0.502709 -0.426996
5 N 1 Z 0.000000 0.142918 0.000000 0.000000 0.000000
6 N 2 S 0.000000 0.011050 0.000000 0.000000 0.000000
7 N 2 S 0.000000 -0.055893 0.000000 0.000000 0.000000
8 N 2 X 0.000000 -0.033015 0.000000 0.000000 0.000000
9 N 2 Y 0.437293 0.000000 -0.582661 -0.502709 0.426996
10 N 2 Z 0.000000 0.142918 0.000000 0.000000 0.000000
11 C 3 S 0.000000 0.012487 0.000000 0.000000 0.000000
12 C 3 S 0.000000 -0.039211 0.000000 0.000000 0.000000
13 C 3 X 0.000000 0.105636 0.000000 0.000000 0.000000
14 C 3 Y 0.362531 0.000000 0.302896 0.184857 0.666591
15 C 3 Z 0.000000 -0.289662 0.000000 0.000000 0.000000
16 C 4 S 0.000000 0.012487 0.000000 0.000000 0.000000
17 C 4 S 0.000000 -0.039211 0.000000 0.000000 0.000000
18 C 4 X 0.000000 -0.105636 0.000000 0.000000 0.000000
19 C 4 Y 0.362531 0.000000 -0.302896 0.184857 -0.666591
20 C 4 Z 0.000000 -0.289662 0.000000 0.000000 0.000000
21 C 5 S 0.000000 0.003715 0.000000 0.000000 0.000000
22 C 5 S 0.000000 0.005600 0.000000 0.000000 0.000000
23 C 5 X 0.000000 0.000000 0.000000 0.000000 0.000000
24 C 5 Y 0.325958 0.000000 0.000000 0.647611 0.000000
25 C 5 Z 0.000000 0.446161 0.000000 0.000000 0.000000
26 H 6 S 0.000000 -0.066833 0.000000 0.000000 0.000000
27 H 7 S 0.000000 -0.066833 0.000000 0.000000 0.000000
28 H 8 S 0.000000 0.269259 0.000000 0.000000 0.000000
29 H 9 S 0.000000 0.269259 0.000000 0.000000 0.000000
30 H 10 S 0.000000 0.403595 0.000000 0.000000 0.000000
31 H 11 S 0.000000 0.123579 0.000000 0.000000 0.000000
32 H 12 S 0.000000 0.123579 0.000000 0.000000 0.000000
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -622.2461877067
TWO ELECTRON ENERGY = 232.3658319988
NUCLEAR REPULSION ENERGY = 164.1314703943
------------------
TOTAL ENERGY = -225.7488853135
ELECTRON-ELECTRON POTENTIAL ENERGY = 232.3658319988
NUCLEUS-ELECTRON POTENTIAL ENERGY = -846.5375156138
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 164.1314703943
------------------
TOTAL POTENTIAL ENERGY = -450.0402132207
TOTAL KINETIC ENERGY = 224.2913279072
VIRIAL RATIO (V/T) = 2.0064985009
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -157.5145234737
BARE H ENERGY= -622.2461877067
ELECTRONIC ENERGY = -389.8803555902
KINETIC ENERGY= 224.2913279072
N-N REPULSION= 164.1314703943
TOTAL ENERGY= -225.7488851958
SIGMA PART(1+2)= -351.9153496040
(K,V1,2)= 214.9517556194 -764.9214820224 198.0543767990
PI PART(1+2)= -37.9650059862
(K,V1,2)= 9.3395722878 -81.6160335914 34.3114553174
SIGMA SKELETON, ERROR= -187.7838792097 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 1.000853 1.000848 -0.000297 -0.000300 0.000001
2 1.000853 1.000848 -0.000297 -0.000300 0.000001
3 -0.000361 -0.000355 1.000873 1.001034 -0.000861
4 -0.000361 -0.000355 1.000873 1.001034 -0.000861
5 0.000002 0.000001 -0.000600 -0.000860 2.002347
6 -0.000247 -0.000247 -0.000002 -0.000002 0.000000
7 -0.000247 -0.000247 -0.000002 -0.000002 0.000000
8 0.000001 0.000001 -0.000271 -0.000299 0.000000
9 0.000001 0.000001 -0.000271 -0.000299 0.000000
10 0.000000 0.000000 0.000000 -0.000001 -0.000625
11 -0.000247 -0.000247 -0.000002 -0.000002 0.000000
12 -0.000247 -0.000247 -0.000002 -0.000002 0.000000
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.570514 0.640771 0.169241 0.298491 0.208178
2 0.570514 0.640771 0.169241 0.298491 0.208178
3 0.270673 0.232476 0.374469 0.383113 0.325428
4 0.270673 0.232476 0.374469 0.383113 0.325428
5 0.108288 0.029088 0.677851 0.197780 0.290680
6 0.042436 0.050928 0.035156 0.063607 0.000105
7 0.042436 0.050928 0.035156 0.063607 0.000105
8 0.015398 0.010696 0.028255 0.093220 0.154670
9 0.015398 0.010696 0.028255 0.093220 0.154670
10 0.006942 0.000000 0.059659 0.000000 0.151643
11 0.043365 0.050584 0.024124 0.062678 0.090458
12 0.043365 0.050584 0.024124 0.062678 0.090458
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.402396 0.464620 0.386402 0.338831 0.188243
2 0.402396 0.464620 0.386402 0.338831 0.188243
3 0.234478 0.274715 0.050653 0.386706 0.321950
4 0.234478 0.274715 0.050653 0.386706 0.321950
5 0.130701 0.104897 0.312683 0.105448 0.394196
6 0.118416 0.162448 0.107664 0.020060 0.110684
7 0.118416 0.162448 0.107664 0.020060 0.110684
8 0.071181 0.031443 0.029512 0.129225 0.113419
9 0.071181 0.031443 0.029512 0.129225 0.113419
10 0.000000 0.014998 0.220914 0.000000 0.000000
11 0.108178 0.006827 0.158969 0.072455 0.068607
12 0.108178 0.006827 0.158969 0.072455 0.068607
16 17 18 19
2.000000 2.000000 2.000000 2.000000
1 0.451949 0.048269 0.750189 0.456041
2 0.451949 0.048269 0.750189 0.456041
3 0.385478 0.222797 0.249811 0.084311
4 0.385478 0.222797 0.249811 0.084311
5 0.325147 0.547489 0.000000 0.919296
6 0.000000 0.008525 0.000000 0.000000
7 0.000000 0.008525 0.000000 0.000000
8 0.000000 0.184485 0.000000 0.000000
9 0.000000 0.184485 0.000000 0.000000
10 0.000000 0.447773 0.000000 0.000000
11 0.000000 0.038292 0.000000 0.000000
12 0.000000 0.038292 0.000000 0.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99702 1.99305
2 N 1 S 1.44435 1.22596
3 N 1 X 1.11160 1.12743
4 N 1 Y 1.65818 1.65200
5 N 1 Z 1.16410 1.16267
6 N 2 S 1.99702 1.99305
7 N 2 S 1.44435 1.22596
8 N 2 X 1.11160 1.12743
9 N 2 Y 1.65818 1.65200
10 N 2 Z 1.16410 1.16267
11 C 3 S 1.99608 1.98983
12 C 3 S 1.13721 1.03521
13 C 3 X 0.91147 0.96957
14 C 3 Y 0.71960 0.72672
15 C 3 Z 1.03302 1.05112
16 C 4 S 1.99608 1.98983
17 C 4 S 1.13721 1.03521
18 C 4 X 0.91147 0.96957
19 C 4 Y 0.71960 0.72672
20 C 4 Z 1.03302 1.05112
21 C 5 S 1.99542 1.98906
22 C 5 S 1.07264 0.98311
23 C 5 X 0.85662 0.93754
24 C 5 Y 1.24444 1.24255
25 C 5 Z 0.97531 1.01190
26 H 6 S 0.71953 0.79979
27 H 7 S 0.71953 0.79979
28 H 8 S 0.86093 0.91187
29 H 9 S 0.86093 0.91187
30 H 10 S 0.90130 0.93885
31 H 11 S 0.72404 0.80327
32 H 12 S 0.72404 0.80327
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2605211
2 -0.0000040 6.2605211
3 0.4601830 0.0008410 4.5619627
4 0.0008410 0.4601830 -0.0302355 4.5619627
5 -0.0276759 -0.0276759 0.5047061 0.5047061 4.8410700
6 0.3554246 0.0000007 -0.0304872 -0.0000671 0.0019438
7 0.0000007 0.3554246 -0.0000671 -0.0304872 0.0019438
8 -0.0244594 0.0000235 0.3894356 -0.0046985 -0.0229973
9 0.0000235 -0.0244594 -0.0046985 0.3894356 -0.0229973
10 -0.0027137 -0.0027137 -0.0238339 -0.0238339 0.3986546
11 -0.0000005 0.3530994 0.0000051 -0.0304247 -0.0036214
12 0.3530994 -0.0000005 -0.0304247 0.0000051 -0.0036214
6 7 8 9 10
6 0.4244387
7 -0.0000001 0.4244387
8 -0.0050421 -0.0000029 0.5239076
9 -0.0000029 -0.0050421 0.0003856 0.5239076
10 0.0000412 0.0000412 0.0017688 0.0017688 0.5504553
11 0.0000001 -0.0267194 0.0000015 0.0026126 0.0008342
12 -0.0267194 0.0000001 0.0026126 0.0000015 0.0008342
11 12
11 0.4282535
12 -0.0000005 0.4282535
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.375240 -0.375240 7.161120 -0.161120
2 N 7.375240 -0.375240 7.161120 -0.161120
3 C 5.797386 0.202614 5.772446 0.227554
4 C 5.797386 0.202614 5.772446 0.227554
5 C 6.144435 -0.144435 6.164155 -0.164155
6 H 0.719530 0.280470 0.799790 0.200210
7 H 0.719530 0.280470 0.799790 0.200210
8 H 0.860935 0.139065 0.911873 0.088127
9 H 0.860935 0.139065 0.911873 0.088127
10 H 0.901303 0.098697 0.938855 0.061145
11 H 0.724040 0.275960 0.803266 0.196734
12 H 0.724040 0.275960 0.803266 0.196734
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND BOND BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER
1 3 1.336 1.387 1 5 2.416 0.072 1 6 1.020 0.892
1 12 1.020 0.893 2 4 1.336 1.387 2 5 2.416 0.072
2 7 1.020 0.892 2 11 1.020 0.893 3 4 2.399 0.052
3 5 1.389 1.383 3 8 1.093 0.953 4 5 1.389 1.383
4 9 1.093 0.953 5 10 1.074 0.965
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 N 3.324 3.324 0.000
2 N 3.324 3.324 0.000
3 C 3.851 3.851 0.000
4 C 3.851 3.851 0.000
5 C 3.898 3.898 0.000
6 H 0.921 0.921 0.000
7 H 0.921 0.921 0.000
8 H 0.981 0.981 0.000
9 H 0.981 0.981 0.000
10 H 0.990 0.990 0.000
11 H 0.924 0.924 0.000
12 H 0.924 0.924 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.030177 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.585043 0.585043
...... END OF PROPERTY EVALUATION ......
CI, STO-6G, 6 p-Elektronen
THE 9 LOWEST DIAGONAL ELEMENTS OF THE HAMILTONIAN ARE
-225.7488853 (CSF 1) -225.4743113 (CSF 170) -225.3610812 (CSF 172)
-225.3328529 (CSF 174) -225.2954089 (CSF 156) -225.2928171 (CSF 171)
-225.2894560 (CSF 175) -225.2687811 (CSF 173) -225.2222276 (CSF 142)
ITER. NO.JUST IMPROVED ENERGY AND STATE
0 30(MAX.TOL.STATE) -225.748885312 1 -225.479157428 2 -225.380660951 3
1 3 0.17435656 2 -225.748885312 1 -225.507644827 2 -225.385906008 3
2 3 0.04167711 3 -225.748885312 1 -225.507814059 2 -225.386353200 3
3 3 0.01465378 3 -225.748885312 1 -225.507819160 2 -225.386411972 3
4 3 0.00653740 3 -225.748885312 1 -225.507819286 2 -225.386419000 3
5 2 0.00114457 3 -225.748885312 1 -225.507819289 2 -225.386419599 3
6 2 0.00059209 3 -225.748885312 1 -225.507819290 2 -225.386419666 3
7 2 0.00019849 3 -225.748885312 1 -225.507819290 2 -225.386419675 3
8 1 0.00007013 3 -225.748885312 1 -225.507819290 2 -225.386419676 3
9 1 0.00002994 3 -225.748885312 1 -225.507819290 2 -225.386419677 3
10 1 0.00001060 3 -225.748885312 1 -225.507819290 2 -225.386419677 3
11 1 0.00000305 3 -225.748885312 1 -225.507819290 2 -225.386419677 3
SOLUTION FOUND WITH INDIRECT METHOD
STATE # 1 ENERGY = -225.748885312
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
1 1.000000 222222222222220000000000000
STATE # 2 ENERGY = -225.507819290
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
34 0.064041 222222222122220000000000100
63 -0.068375 222222221222220000000010000
78 0.050961 222222212222220000000100000
157 -0.104855 222222222222120100000000000
170 0.977984 222222222222211000000000000
STATE # 3 ENERGY = -225.386419677
CSF COEF OCCUPANCY (IGNORING CORE)
--- ---- --------- --------- -----
160 0.269344 222222222122220100000000000
161 -0.074371 222222221222220100000000000
164 0.099604 222221222222220100000000000
172 0.941961 222222222221221000000000000
176 0.058818 222222212222221000000000000
179 0.122369 222212222222221000000000000
181 0.052037 221222222222221000000000000
...... END OF CI-MATRIX DIAGONALIZATION ......
16 17 18 19 20
2.0000 2.0000 2.0000 2.0000 0.0000
1 N 1 S 0.000003 0.000070 0.704333 0.000067 0.000000
2 N 1 S 0.000093 -0.002695 0.014230 -0.002673 0.000000
3 N 1 X -0.000062 0.001580 -0.000545 0.001553 0.000000
4 N 1 Y 0.000000 0.000000 0.000000 0.000000 -0.426996
5 N 1 Z 0.000108 0.000665 -0.000264 0.000674 0.000000
6 N 2 S 0.000003 0.000070 0.704333 -0.000067 0.000000
7 N 2 S 0.000093 -0.002695 0.014230 0.002673 0.000000
8 N 2 X 0.000062 -0.001580 0.000545 0.001553 0.000000
9 N 2 Y 0.000000 0.000000 0.000000 0.000000 0.426996
10 N 2 Z 0.000108 0.000665 -0.000264 -0.000674 0.000000
11 C 3 S -0.001828 0.703953 0.000226 0.703926 0.000000
12 C 3 S -0.004855 0.015266 -0.002843 0.015275 0.000000
13 C 3 X 0.002818 0.000297 -0.002089 0.000213 0.000000
14 C 3 Y 0.000000 0.000000 0.000000 0.000000 0.666591
15 C 3 Z -0.001554 0.000381 -0.000939 0.000399 0.000000
16 C 4 S -0.001828 0.703953 0.000226 -0.703926 0.000000
17 C 4 S -0.004855 0.015266 -0.002843 -0.015275 0.000000
18 C 4 X -0.002818 -0.000297 0.002089 0.000213 0.000000
19 C 4 Y 0.000000 0.000000 0.000000 0.000000 -0.666591
20 C 4 Z -0.001554 0.000381 -0.000939 -0.000399 0.000000
21 C 5 S 0.995140 0.003155 -0.000010 0.000000 0.000000
22 C 5 S 0.023952 -0.005759 0.000108 0.000000 0.000000
23 C 5 X 0.000000 0.000000 0.000000 -0.003447 0.000000
24 C 5 Y 0.000000 0.000000 0.000000 0.000000 0.000000
25 C 5 Z -0.001371 0.001775 0.000264 0.000000 0.000000
26 H 6 S 0.000004 -0.000205 -0.002772 -0.000200 0.000000
27 H 7 S 0.000004 -0.000205 -0.002772 0.000200 0.000000
28 H 8 S 0.000005 -0.002989 0.000089 -0.002880 0.000000
29 H 9 S 0.000005 -0.002989 0.000089 0.002880 0.000000
30 H 10 S -0.004353 -0.000067 -0.000048 0.000000 0.000000
31 H 11 S -0.000131 -0.000218 -0.002766 0.000208 0.000000
32 H 12 S -0.000131 -0.000218 -0.002766 -0.000208 0.000000
-----------------------------
properties for the CI density
-----------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -622.2461876773
TWO ELECTRON ENERGY = 232.3658319711
NUCLEAR REPULSION ENERGY = 164.1314703943
------------------
TOTAL ENERGY = -225.7488853119
ELECTRON-ELECTRON POTENTIAL ENERGY = 232.3658319711
NUCLEUS-ELECTRON POTENTIAL ENERGY = -846.5375156129
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 164.1314703943
------------------
TOTAL POTENTIAL ENERGY = -450.0402132475
TOTAL KINETIC ENERGY = 224.2913279356
VIRIAL RATIO (V/T) = 2.0064985008
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.456041 0.451949 0.570514 0.640771 0.169241
2 0.456041 0.451949 0.570514 0.640771 0.169241
3 0.084311 0.385478 0.270673 0.232476 0.374469
4 0.084311 0.385478 0.270673 0.232476 0.374469
5 0.919297 0.325147 0.108288 0.029088 0.677851
6 0.000000 0.000000 0.042436 0.050928 0.035156
7 0.000000 0.000000 0.042436 0.050928 0.035156
8 0.000000 0.000000 0.015398 0.010696 0.028255
9 0.000000 0.000000 0.015398 0.010696 0.028255
10 0.000000 0.000000 0.006942 0.000000 0.059659
11 0.000000 0.000000 0.043365 0.050584 0.024124
12 0.000000 0.000000 0.043365 0.050584 0.024124
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.298491 0.208178 0.402396 0.750189 0.386402
2 0.298491 0.208178 0.402396 0.750189 0.386402
3 0.383113 0.325428 0.234478 0.249811 0.050653
4 0.383113 0.325428 0.234478 0.249811 0.050653
5 0.197780 0.290680 0.130701 0.000000 0.312683
6 0.063607 0.000105 0.118416 0.000000 0.107664
7 0.063607 0.000105 0.118416 0.000000 0.107664
8 0.093220 0.154670 0.071181 0.000000 0.029512
9 0.093220 0.154670 0.071181 0.000000 0.029512
10 0.000000 0.151643 0.000000 0.000000 0.220914
11 0.062678 0.090458 0.108178 0.000000 0.158969
12 0.062678 0.090458 0.108178 0.000000 0.158969
11 12 13 14 15
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.188243 0.048269 0.464620 0.338831 1.000853
2 0.188243 0.048269 0.464620 0.338831 1.000853
3 0.321950 0.222797 0.274715 0.386706 -0.000361
4 0.321950 0.222797 0.274715 0.386706 -0.000361
5 0.394196 0.547490 0.104897 0.105448 0.000002
6 0.110684 0.008525 0.162448 0.020060 -0.000247
7 0.110684 0.008525 0.162448 0.020060 -0.000247
8 0.113418 0.184485 0.031443 0.129225 0.000001
9 0.113418 0.184485 0.031443 0.129225 0.000001
10 0.000000 0.447774 0.014998 0.000000 0.000000
11 0.068607 0.038292 0.006827 0.072455 -0.000247
12 0.068607 0.038292 0.006827 0.072455 -0.000247
16 17 18 19 20
2.000000 2.000000 2.000000 2.000000 0.000000
1 0.000001 -0.000300 1.000848 -0.000297 0.000000
2 0.000001 -0.000300 1.000848 -0.000297 0.000000
3 -0.000861 1.001034 -0.000355 1.000873 0.000000
4 -0.000861 1.001034 -0.000355 1.000873 0.000000
5 2.002347 -0.000860 0.000001 -0.000600 0.000000
6 0.000000 -0.000002 -0.000247 -0.000002 0.000000
7 0.000000 -0.000002 -0.000247 -0.000002 0.000000
8 0.000000 -0.000299 0.000001 -0.000271 0.000000
9 0.000000 -0.000299 0.000001 -0.000271 0.000000
10 -0.000625 -0.000001 0.000000 0.000000 0.000000
11 0.000000 -0.000002 -0.000247 -0.000002 0.000000
12 0.000000 -0.000002 -0.000247 -0.000002 0.000000
21 22 23 24 25
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
9 0.000000 0.000000 0.000000 0.000000 0.000000
10 0.000000 0.000000 0.000000 0.000000 0.000000
11 0.000000 0.000000 0.000000 0.000000 0.000000
12 0.000000 0.000000 0.000000 0.000000 0.000000
26 27 28 29 30
0.000000 0.000000 0.000000 0.000000 0.000000
1 0.000000 0.000000 0.000000 0.000000 0.000000
2 0.000000 0.000000 0.000000 0.000000 0.000000
3 0.000000 0.000000 0.000000 0.000000 0.000000
4 0.000000 0.000000 0.000000 0.000000 0.000000
5 0.000000 0.000000 0.000000 0.000000 0.000000
6 0.000000 0.000000 0.000000 0.000000 0.000000
7 0.000000 0.000000 0.000000 0.000000 0.000000
8 0.000000 0.000000 0.000000 0.000000 0.000000
9 0.000000 0.000000 0.000000 0.000000 0.000000
10 0.000000 0.000000 0.000000 0.000000 0.000000
11 0.000000 0.000000 0.000000 0.000000 0.000000
12 0.000000 0.000000 0.000000 0.000000 0.000000
31 32
0.000000 0.000000
1 0.000000 0.000000
2 0.000000 0.000000
3 0.000000 0.000000
4 0.000000 0.000000
5 0.000000 0.000000
6 0.000000 0.000000
7 0.000000 0.000000
8 0.000000 0.000000
9 0.000000 0.000000
10 0.000000 0.000000
11 0.000000 0.000000
12 0.000000 0.000000
WARNING! CI POPULATIONS SHOWN ABOVE ARE FOR THE NATURAL ORBITALS.
IGNORE THE ABOVE DATA FOR CI FUNCTIONS WHICH ARE NOT OF -FORS- TYPE.
THE FOLLOWING POPULATIONS ARE CORRECT FOR ANY CI WAVEFUNCTION.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 N 1 S 1.99702 1.99305
2 N 1 S 1.44435 1.22596
3 N 1 X 1.11160 1.12743
4 N 1 Y 1.65818 1.65200
5 N 1 Z 1.16410 1.16267
6 N 2 S 1.99702 1.99305
7 N 2 S 1.44435 1.22596
8 N 2 X 1.11160 1.12743
9 N 2 Y 1.65818 1.65200
10 N 2 Z 1.16410 1.16267
11 C 3 S 1.99608 1.98983
12 C 3 S 1.13721 1.03521
13 C 3 X 0.91147 0.96957
14 C 3 Y 0.71960 0.72672
15 C 3 Z 1.03302 1.05112
16 C 4 S 1.99608 1.98983
17 C 4 S 1.13721 1.03521
18 C 4 X 0.91147 0.96957
19 C 4 Y 0.71960 0.72672
20 C 4 Z 1.03302 1.05112
21 C 5 S 1.99542 1.98906
22 C 5 S 1.07264 0.98311
23 C 5 X 0.85662 0.93754
24 C 5 Y 1.24444 1.24255
25 C 5 Z 0.97531 1.01190
26 H 6 S 0.71953 0.79979
27 H 7 S 0.71953 0.79979
28 H 8 S 0.86093 0.91187
29 H 9 S 0.86093 0.91187
30 H 10 S 0.90130 0.93885
31 H 11 S 0.72404 0.80327
32 H 12 S 0.72404 0.80327
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1 2 3 4 5
1 6.2605212
2 -0.0000040 6.2605212
3 0.4601829 0.0008410 4.5619627
4 0.0008410 0.4601829 -0.0302355 4.5619627
5 -0.0276759 -0.0276759 0.5047061 0.5047061 4.8410699
6 0.3554246 0.0000007 -0.0304872 -0.0000671 0.0019438
7 0.0000007 0.3554246 -0.0000671 -0.0304872 0.0019438
8 -0.0244594 0.0000235 0.3894356 -0.0046985 -0.0229973
9 0.0000235 -0.0244594 -0.0046985 0.3894356 -0.0229973
10 -0.0027137 -0.0027137 -0.0238339 -0.0238339 0.3986546
11 -0.0000005 0.3530994 0.0000051 -0.0304247 -0.0036214
12 0.3530994 -0.0000005 -0.0304247 0.0000051 -0.0036214
6 7 8 9 10
6 0.4244387
7 -0.0000001 0.4244387
8 -0.0050421 -0.0000029 0.5239076
9 -0.0000029 -0.0050421 0.0003856 0.5239076
10 0.0000412 0.0000412 0.0017688 0.0017688 0.5504553
11 0.0000001 -0.0267194 0.0000015 0.0026126 0.0008342
12 -0.0267194 0.0000001 0.0026126 0.0000015 0.0008342
11 12
11 0.4282535
12 -0.0000005 0.4282535
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 N 7.375240 -0.375240 7.161120 -0.161120
2 N 7.375240 -0.375240 7.161120 -0.161120
3 C 5.797386 0.202614 5.772446 0.227554
4 C 5.797386 0.202614 5.772446 0.227554
5 C 6.144435 -0.144435 6.164155 -0.164155
6 H 0.719530 0.280470 0.799790 0.200210
7 H 0.719530 0.280470 0.799790 0.200210
8 H 0.860935 0.139065 0.911873 0.088127
9 H 0.860935 0.139065 0.911873 0.088127
10 H 0.901303 0.098697 0.938855 0.061145
11 H 0.724040 0.275960 0.803266 0.196734
12 H 0.724040 0.275960 0.803266 0.196734
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.030177 1.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 -0.585044 0.585044
...... END OF PROPERTY EVALUATION ......
| Seitenanfang |