SiO2-Molekül
Input
!
! SiO2
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=0 RUNTYP=OPTIMIZE COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
SiO2
CNH 2
Si 14.0 0.00 0.00 0.00
O 8.0 0.00 1.50 0.00
$END
Output
TOTAL NUMBER OF SHELLS = 7
TOTAL NUMBER OF BASIS FUNCTIONS = 19
NUMBER OF ELECTRONS = 30
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 15
NUMBER OF OCCUPIED ORBITALS (BETA ) = 15
TOTAL NUMBER OF ATOMS = 3
NSERCH= 4 ENERGY= -436.8282543
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX
DE/DY DE/DZ
--------------------------------------------------------------
1 SI 14.0 0.0000000 0.0000000
0.0000000
2 O 8.0 0.0000000
-0.0000152 0.0000000
3 O 8.0 0.0000000
0.0000152 0.0000000
MAXIMUM GRADIENT = 0.0000152 RMS GRADIENT = 0.0000071
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
SiO2
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
SI 14.0 0.0000000000 0.0000000000 0.0000000000
O 8.0 0.0000000000 1.5734603121 0.0000000000
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X
Y
Z
------------------------------------------------------------
SI 14.0 0.0000000000 0.0000000000
0.0000000000
O 8.0 0.0000000000 -1.5734603121
0.0000000000
O 8.0 0.0000000000
1.5734603121 0.0000000000
z ist die C2-Achse, y ist die
Bindungsachse
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
SI
O O
1 SI 0.0000000
1.5734603 * 1.5734603
*
2 O 1.5734603 * 0.0000000
3.1469206
3 O 1.5734603 * 3.1469206
0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 86.0964682517
ELECTRONIC ENERGY = -522.9247225092
TOTAL ENERGY = -436.8282542574
------------------
MOLECULAR ORBITALS
------------------
1 2
3 4
5
-68.9242 -20.4745 -20.4742 -6.1024
-4.1574
AG BU
AG AG
BU
1 SI 1 S 0.997358 0.000000 -0.000075
-0.332809 0.000000
2 SI 1 S 0.008412 0.000000
0.000097 1.044547 0.000000
3 SI 1 X 0.000000 0.000000
0.000000 0.000000 0.000000
4 SI 1 Y 0.000000 -0.000683 0.000000
0.000000 0.983114
5 SI 1 Z 0.000000 0.000000
0.000000 0.000000 0.000000
6 SI 1 S -0.001073 0.000000 -0.003670
0.032619 0.000000
7 SI 1 X 0.000000 0.000000
0.000000 0.000000 0.000000
8 SI 1 Y 0.000000 0.006921
0.000000 0.000000 0.077178
9 SI 1 Z 0.000000 0.000000
0.000000 0.000000 0.000000
10 O 2 S -0.000016 0.704717 0.704912
-0.000037 0.000068
11 O 2 S 0.000216 0.011753
0.009776 -0.004596 0.018627
12 O 2 X 0.000000 0.000000
0.000000 0.000000 0.000000
13 O 2 Y 0.000031 0.002632
0.001584 0.000608 0.000825
14 O 2 Z 0.000000 0.000000
0.000000 0.000000 0.000000
15 O 3 S -0.000016 -0.704717 0.704912
-0.000037 -0.000068
16 O 3 S 0.000216 -0.011753 0.009776
-0.004596 -0.018627
17 O 3 X 0.000000 0.000000
0.000000 0.000000 0.000000
18 O 3 Y -0.000031 0.002632 -0.001584
-0.000608 0.000825
19 O 3 Z 0.000000 0.000000
0.000000 0.000000 0.000000
6 7
8 9
10
-4.1539 -4.1539 -1.2250
-1.2071 -0.5271
BU AU
AG BU
AG
1 SI 1 S 0.000000 0.000000
0.029873 0.000000 0.058786
2 SI 1 S 0.000000 0.000000 -0.112329
0.000000 -0.204331
3 SI 1 X 0.986744 0.000000
0.000000 0.000000 0.000000
4 SI 1 Y 0.000000 0.000000
0.000000 0.102028 0.000000
5 SI 1 Z 0.000000 0.986744
0.000000 0.000000 0.000000
6 SI 1 S 0.000000 0.000000
0.222093 0.000000 0.753230
7 SI 1 X 0.057319 0.000000
0.000000 0.000000 0.000000
8 SI 1 Y 0.000000 0.000000
0.000000 -0.168277 0.000000
9 SI 1 Z 0.000000 0.057319
0.000000 0.000000 0.000000
10 O 2 S 0.000000 0.000000 -0.160925
-0.164526 0.077701
11 O 2 S 0.000000 0.000000
0.631107 0.636753 -0.428751
12 O 2 X -0.004359 0.000000 0.000000
0.000000 0.000000
13 O 2 Y 0.000000 0.000000
0.093360 0.080281 0.371305
14 O 2 Z 0.000000 -0.004359 0.000000
0.000000 0.000000
15 O 3 S 0.000000 0.000000 -0.160925
0.164526 0.077701
16 O 3 S 0.000000 0.000000
0.631107 -0.636753 -0.428751
17 O 3 X -0.004359 0.000000 0.000000
0.000000 0.000000
18 O 3 Y 0.000000 0.000000 -0.093360
0.080281 -0.371305
19 O 3 Z 0.000000 -0.004359 0.000000
0.000000 0.000000
HOMO
11 12
13 14
15
-0.4467 -0.4467 -0.3924
-0.3226 -0.3226
AU BU
BU AG
BG
1 SI 1 S 0.000000 0.000000
0.000000 0.000000 0.000000
2 SI 1 S 0.000000 0.000000
0.000000 0.000000 0.000000
3 SI 1 X 0.000000 -0.149411 0.000000
0.000000 0.000000
4 SI 1 Y 0.000000 0.000000
0.160015 0.000000 0.000000
5 SI 1 Z -0.149411 0.000000 0.000000
0.000000 0.000000
6 SI 1 S 0.000000 0.000000
0.000000 0.000000 0.000000
7 SI 1 X 0.000000
0.527119 0.000000
0.000000 0.000000
8 SI 1 Y 0.000000 0.000000 -0.483349
0.000000 0.000000
9 SI 1 Z
0.527119 0.000000 0.000000 0.000000
0.000000
10 O 2 S 0.000000 0.000000
0.058430 0.000000 0.000000
11 O 2 S 0.000000 0.000000 -0.360652
0.000000 0.000000
12 O 2 X 0.000000
0.528357 0.000000
0.707236 0.000000
13 O 2 Y 0.000000 0.000000
0.534095 0.000000 0.000000
14 O 2 Z
0.528357
0.000000 0.000000 0.000000
0.707236
15 O 3 S 0.000000 0.000000
-0.058430 0.000000 0.000000
16 O 3 S 0.000000 0.000000
0.360652 0.000000 0.000000
17 O 3 X 0.000000
0.528357 0.000000
-0.707236 0.000000
18 O 3 Y 0.000000 0.000000
0.534095 0.000000 0.000000
19 O 3 Z
0.528357
0.000000 0.000000 0.000000
-0.707236
Die
bindenden
p-MO's sind MO11 und MO12.
MO11
MO12
MO13
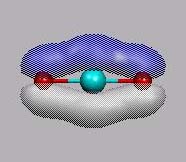
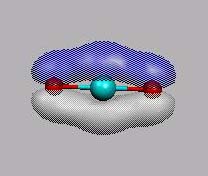
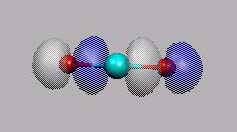
MO11 und MO12 sind die beiden
bindenden p-MO's
(pz- und px-Kombination).
Man beachte, dass die äußeren px-AO's alle das gleiche Vorzeichnen
haben, daraus könnte man den Schluss ziehen, dass es ein antibindendes MO ist.
Man muss die Symmetrieeigenschaft (Bu) berücksichtigen.
MO14
MO15 = HOMO
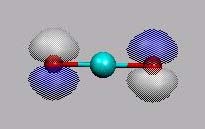
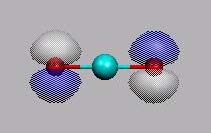
LUMO
16 17
18 19
0.1860 0.1860 0.1909
0.7611
BU AU
AG BU
1 SI 1 S 0.000000 0.000000
0.045652 0.000000
2 SI 1 S 0.000000 0.000000 -0.138471
0.000000
3 SI 1 X -0.223084 0.000000 0.000000
0.000000
4 SI 1 Y 0.000000 0.000000
0.000000 -0.251157
5 SI 1 Z 0.000000 -0.223084 0.000000
0.000000
6 SI 1 S 0.000000 0.000000
0.908692 0.000000
7 SI 1 X 0.905234 0.000000
0.000000 0.000000
8 SI 1 Y 0.000000 0.000000
0.000000 1.401284
9 SI 1 Z 0.000000 0.905234
0.000000 0.000000
10 O 2 S 0.000000 0.000000
0.040890 -0.079687
11 O 2 S 0.000000 0.000000 -0.293916
0.665471
12 O 2 X -0.497634 0.000000 0.000000
0.000000
13 O 2 Y 0.000000 0.000000 -0.638871
0.593428
14 O 2 Z 0.000000 -0.497634 0.000000
0.000000
15 O 3 S 0.000000 0.000000
0.040890 0.079687
16 O 3 S 0.000000 0.000000 -0.293916
-0.665471
17 O 3 X -0.497634 0.000000 0.000000
0.000000
18 O 3 Y 0.000000 0.000000
0.638871 0.593428
19 O 3 Z 0.000000 -0.497634 0.000000
0.000000
LUMO

------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -779.1878498439
TWO ELECTRON ENERGY = 256.2631273348
NUCLEAR REPULSION ENERGY = 86.0964682517
------------------
TOTAL ENERGY = -436.8282542574
ELECTRON-ELECTRON POTENTIAL ENERGY = 256.2631273348
NUCLEUS-ELECTRON POTENTIAL ENERGY = -1216.4239644801
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 86.0964682517
------------------
TOTAL POTENTIAL ENERGY = -874.0643688936
TOTAL KINETIC ENERGY = 437.2361146362
VIRIAL RATIO (V/T) = 1.9990671851
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -266.6615946454
BARE H ENERGY= -779.1878498439
ELECTRONIC ENERGY = -522.9247222447
KINETIC ENERGY= 437.2361146362
N-N REPULSION= 86.0964682517
TOTAL ENERGY= -436.8282539929
SIGMA PART(1+2)= -409.7803437640
(K,V1,2)= 374.1621161984 -946.7541018530 162.8116418906
PI PART(1+2)= -113.1443784806
(K,V1,2)= 63.0739984378 -269.6698626271 93.4514857086
SIGMA SKELETON, ERROR= -323.6838755123 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1
2 3
4 5
2.000000 2.000000
2.000000 2.000000 2.000000
1 1.999997 -0.001153 -0.000357
2.000489 2.003026
2 0.000001 1.000576 1.000178
-0.000245 -0.001513
3 0.000001 1.000576 1.000178
-0.000245 -0.001513
6
7 8
9 10
2.000000 2.000000
2.000000 2.000000 2.000000
1 2.000208 2.000208 0.276844
0.252320 0.917629
2 -0.000104 -0.000104 0.861578
0.873840 0.541186
3 -0.000104 -0.000104 0.861578
0.873840 0.541186
11
12 13
14 15
2.000000 2.000000
2.000000 2.000000 2.000000
1 0.709214 0.709214 0.410052
0.000000 0.000000
2 0.645393 0.645393 0.794974
1.000000 1.000000
3 0.645393 0.645393 0.794974
1.000000 1.000000
Die bindenden
p-MO's
sind MO11 und MO12.
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 SI 1 S 1.99994 1.99951
2 SI 1 S 1.99637 1.99256
3 SI 1 X 1.98040 1.96758
4 SI 1 Y 1.98063 1.97438
5 SI 1 Z 1.98040 1.96758
6 SI 1 S 1.19829 1.21440
7 SI 1 X 0.72902
0.74557
8 SI 1 Y 0.68361 0.81293
9 SI 1 Z 0.72902
0.74557
10 O 2 S 1.99921 1.99871
11 O 2 S 1.94939 1.84210
12 O 2 X 1.64529 1.64342
13 O 2 Y 1.12199 1.16230
14 O 2 Z 1.64529 1.64342
15 O 3 S 1.99921 1.99871
16 O 3 S 1.94939 1.84210
17 O 3 X 1.64529 1.64342
18 O 3 Y 1.12199 1.16230
19 O 3 Z 1.64529 1.64342
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1
2 3
1 12.7844587
2 0.2466155 8.1154149
3 0.2466155
-0.0008753 8.1154149
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE
LOW.POP. CHARGE
1 SI 13.277690 0.722310 13.420090
0.579910
2 O 8.361155 -0.361155
8.289955 -0.289955
3 O 8.361155 -0.361155
8.289955 -0.289955
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR
DIST ORDER
1 2 1.573
1.840 1 3
1.573 1.840 2 3 3.147 0.349
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 SI 3.681 3.681 0.000
2 O 2.189 2.189 0.000
3 O 2.189 2.189 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......