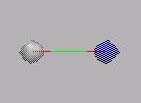
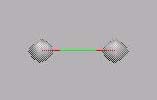

MO1, MO2: Beide Elektronen befinden sich fast ausschließlich am O-Atom (s.
O-Atom). Im MO3 befindet sich das Elektronenpaar am C-Atom.
| Arbeiten mit GAMESS |
Input
!
! CO2
!
$CONTRL SCFTYP=RHF MULT=1 ICHARG=0 RUNTYP=OPTIMIZE COORD=UNIQUE $END
$SYSTEM TIMLIM=10 MEMORY=60000000 $END
$BASIS GBASIS=STO NGAUSS=6 $END
$GUESS GUESS=HUCKEL $END
$DATA
CO2
CNH 2
C 6.0 0.00 0.00 0.00
O 8.0 0.00 1.20 0.00
$END
Output
ATOMIC BASIS SET
----------------
THE CONTRACTED PRIMITIVE FUNCTIONS HAVE BEEN UNNORMALIZED
THE CONTRACTED BASIS FUNCTIONS ARE NOW NORMALIZED TO UNITY
SHELL TYPE PRIM EXPONENT CONTRACTION COEFFICIENTS
C
1 S 1 742.737049 0.929185 ( 0.009164)
1 S 2 136.180025 1.402437 ( 0.049361)
1 S 3 38.098264 1.841991 ( 0.168538)
1 S 4 13.087782 1.817278 ( 0.370563)
1 S 5 5.082369 1.004768 ( 0.416492)
1 S 6 2.093200 0.161650 ( 0.130334)
2 L 7 30.497240 -0.122578 ( -0.013253) 0.384077 ( 0.003760)
2 L 8 6.036200 -0.128975 ( -0.046992) 0.508157 ( 0.037679)
2 L 9 1.876046 -0.038599 ( -0.033785) 0.544234 ( 0.173897)
2 L 10 0.721783 0.139661 ( 0.250242) 0.396426 ( 0.418036)
2 L 11 0.313471 0.177689 ( 0.595117) 0.142381 ( 0.425860)
2 L 12 0.143687 0.040037 ( 0.240706) 0.012825 ( 0.101708)
O
5 S 13 1355.584234 1.459053 ( 0.009164)
5 S 14 248.544886 2.202176 ( 0.049361)
5 S 15 69.533902 2.892385 ( 0.168538)
5 S 16 23.886772 2.853580 ( 0.370563)
5 S 17 9.275933 1.577736 ( 0.416492)
5 S 18 3.820341 0.253831 ( 0.130334)
6 L 19 52.187762 -0.183398 ( -0.013253) 0.751716 ( 0.003760)
6 L 20 10.329320 -0.192968 ( -0.046992) 0.994565 ( 0.037679)
6 L 21 3.210345 -0.057750 ( -0.033785) 1.065175 ( 0.173897)
6 L 22 1.235135 0.208956 ( 0.250242) 0.775885 ( 0.418036)
6 L 23 0.536420 0.265853 ( 0.595117) 0.278668 ( 0.425860)
6 L 24 0.245881 0.059902 ( 0.240706) 0.025102 ( 0.101708)
TOTAL NUMBER OF SHELLS = 6
TOTAL NUMBER OF BASIS FUNCTIONS = 15
NUMBER OF ELECTRONS = 22
CHARGE OF MOLECULE = 0
STATE MULTIPLICITY = 1
NUMBER OF OCCUPIED ORBITALS (ALPHA) = 11
NUMBER OF OCCUPIED ORBITALS (BETA ) = 11
TOTAL NUMBER OF ATOMS = 3
THE NUCLEAR REPULSION ENERGY IS 56.4455732523 NSERCH= 5 ENERGY= -186.8553185
-----------------------
GRADIENT (HARTREE/BOHR)
-----------------------
ATOM ZNUC DE/DX DE/DY DE/DZ
--------------------------------------------------------------
1 C 6.0 0.0000000 0.0000000 0.0000000
2 O 8.0 0.0000000 0.0000002 0.0000000
3 O 8.0 0.0000000 -0.0000002 0.0000000
MAXIMUM GRADIENT = 0.0000002 RMS GRADIENT = 0.0000001
1 ***** EQUILIBRIUM GEOMETRY LOCATED *****
CO2
COORDINATES OF SYMMETRY UNIQUE ATOMS (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C 6.0 0.0000000000 0.0000000000 0.0000000000
O 8.0 0.0000000000 1.1866204525 0.0000000000
COORDINATES OF ALL ATOMS ARE (ANGS)
ATOM CHARGE X Y Z
------------------------------------------------------------
C 6.0 0.0000000000 0.0000000000 0.0000000000
O 8.0 0.0000000000 -1.1866204525 0.0000000000
O 8.0 0.0000000000 1.1866204525 0.0000000000
INTERNUCLEAR DISTANCES (ANGS.)
------------------------------
C O O
1 C 0.0000000 1.1866205 * 1.1866205 *
2 O 1.1866205 * 0.0000000 2.3732409 *
3 O 1.1866205 * 2.3732409 * 0.0000000
* ... LESS THAN 3.000
NUCLEAR ENERGY = 57.0820162072
ELECTRONIC ENERGY = -243.9373347023
TOTAL ENERGY = -186.8553184952
------------------
MOLECULAR ORBITALS
------------------
1 2 3 4 5
-20.6214 -20.6212 -11.4213 -1.4435 -1.3796
BU AG AG AG BU
1 C 1 S 0.000000 0.000340 0.995989 -0.154435 0.000000
2 C 1 S 0.000000 -0.006146 0.017205 0.342005 0.000000
3 C 1 X 0.000000 0.000000 0.000000 0.000000 0.000000
4 C 1 Y 0.007392 0.000000 0.000000 0.000000 -0.303353
5 C 1 Z 0.000000 0.000000 0.000000 0.000000 0.000000
6 O 2 S 0.704741 0.704818 -0.000143 -0.141983 -0.154592
7 O 2 S 0.012212 0.010709 -0.002830 0.522728 0.546659
8 O 2 X 0.000000 0.000000 0.000000 0.000000 0.000000
9 O 2 Y 0.003798 0.002820 -0.000387 0.141783 0.114366
10 O 2 Z 0.000000 0.000000 0.000000 0.000000 0.000000
11 O 3 S -0.704741 0.704818 -0.000143 -0.141983 0.154592
12 O 3 S -0.012212 0.010709 -0.002830 0.522728 -0.546659
13 O 3 X 0.000000 0.000000 0.000000 0.000000 0.000000
14 O 3 Y 0.003798 -0.002820 0.000387 -0.141783 0.114366
15 O 3 Z 0.000000 0.000000 0.000000 0.000000 0.000000
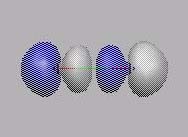
MO1
MO2
MO3
MO1, MO2: Beide Elektronen befinden
sich fast ausschließlich am O-Atom (s.
O-Atom). Im MO3 befindet sich das Elektronenpaar am
C-Atom.
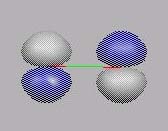
MO4
MO5
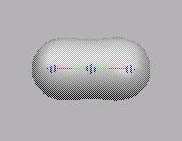

MO4 ist das erste "echte" MO, gebildet aus den
Valenz-AO's. Es enthält eine innere sphärische Knotenebene.
6 7 8 9 10
-0.6886 -0.6273 -0.6273 -0.5910 -0.3964
AG AU BU BU BG
1 C 1 S -0.157721 0.000000 0.000000 0.000000 0.000000
2 C 1 S 0.561135 0.000000 0.000000 0.000000 0.000000
3 C 1 X 0.000000 0.000000
0.556098 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 -0.442853 0.000000
5 C 1 Z 0.000000
0.556098 0.000000 0.000000 0.000000
6 O 2 S 0.101541 0.000000 0.000000 0.075289 0.000000
7 O 2 S -0.520884 0.000000 0.000000 -0.445837 0.000000
8 O 2 X 0.000000 0.000000
0.476290 0.000000 0.000000
9 O 2 Y 0.406086 0.000000 0.000000 0.498912 0.000000
10 O 2 Z 0.000000
0.476290 0.000000 0.000000 0.708885
11 O 3 S 0.101541 0.000000 0.000000 -0.075289 0.000000
12 O 3 S -0.520884 0.000000 0.000000 0.445837 0.000000
13 O 3 X 0.000000 0.000000
0.476290 0.000000 0.000000
14 O 3 Y -0.406086 0.000000 0.000000 0.498912 0.000000
15 O 3 Z 0.000000
0.476290 0.000000 0.000000 -0.708885
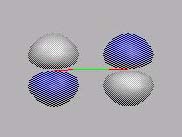
bindende
p-MO's
MO6
MO7
MO8

MO9
MO10
HOMO
11 12 13 14 15
-0.3964 0.2995 0.2995 0.5240 1.2854
AG BU AU AG BU
1 C 1 S 0.000000 0.000000 0.000000 -0.182448 0.000000
2 C 1 S 0.000000 0.000000 0.000000 1.280241 0.000000
3 C 1 X 0.000000 0.893904 0.000000 0.000000 0.000000
4 C 1 Y 0.000000 0.000000 0.000000 0.000000 1.576299
5 C 1 Z 0.000000 0.000000 0.893904 0.000000 0.000000
6 O 2 S 0.000000 0.000000 0.000000 0.070995 -0.084605
7 O 2 S 0.000000 0.000000 0.000000 -0.511975 0.820527
8 O 2 X 0.708885 -0.569684 0.000000 0.000000 0.000000
9 O 2 Y 0.000000 0.000000 0.000000 -0.698340 0.755840
10 O 2 Z 0.000000 0.000000 -0.569684 0.000000 0.000000
11 O 3 S 0.000000 0.000000 0.000000 0.070995 0.084605
12 O 3 S 0.000000 0.000000 0.000000 -0.511975 -0.820527
13 O 3 X -0.708885 -0.569684 0.000000 0.000000 0.000000
14 O 3 Y 0.000000 0.000000 0.000000 0.698340 0.755840
15 O 3 Z 0.000000 0.000000 -0.569684 0.000000 0.000000
MO11
MO12
MO13
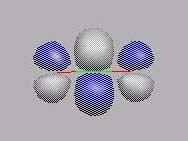

------------------------------
properties for the RHF density
------------------------------
-----------------
ENERGY COMPONENTS
-----------------
WAVEFUNCTION NORMALIZATION = 1.0000000000
ONE ELECTRON ENERGY = -370.2469856717
TWO ELECTRON ENERGY = 126.3096509693
NUCLEAR REPULSION ENERGY = 57.0820162072
------------------
TOTAL ENERGY = -186.8553184952
ELECTRON-ELECTRON POTENTIAL ENERGY = 126.3096509693
NUCLEUS-ELECTRON POTENTIAL ENERGY = -556.7065096413
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 57.0820162072
------------------
TOTAL POTENTIAL ENERGY = -373.3148424648
TOTAL KINETIC ENERGY = 186.4595239696
VIRIAL RATIO (V/T) = 2.0021226833
...... PI ENERGY ANALYSIS ......
ENERGY ANALYSIS:
FOCK ENERGY= -117.6276854280
BARE H ENERGY= -370.2469856717
ELECTRONIC ENERGY = -243.9373355498
KINETIC ENERGY= 186.4595239696
N-N REPULSION= 57.0820162072
TOTAL ENERGY= -186.8553193427
SIGMA PART(1+2)= -199.4136686736
(K,V1,2)= 169.3861157533 -454.6805378284 85.8807534015
PI PART(1+2)= -44.5236668762
(K,V1,2)= 17.0734082163 -102.0259718129 40.4288967204
SIGMA SKELETON, ERROR= -142.3316524665 0.0000000000
MIXED PART= 0.00000E+00 0.00000E+00 0.00000E+00 0.00000E+00
...... END OF PI ENERGY ANALYSIS ......
---------------------------------------
MULLIKEN AND LOWDIN POPULATION ANALYSES
---------------------------------------
MULLIKEN ATOMIC POPULATION IN EACH MOLECULAR ORBITAL
1 2 3 4 5
2.000000 2.000000 2.000000 2.000000 2.000000
1 -0.001522 -0.000746 2.000609 0.539536 0.514994
2 1.000761 1.000373 -0.000305 0.730232 0.742503
3 1.000761 1.000373 -0.000305 0.730232 0.742503
6 7 8 9 10
2.000000 2.000000 2.000000 2.000000 2.000000
1 0.452714 0.853268 0.853268 0.322433 0.000000
2 0.773643 0.573366 0.573366 0.838784 1.000000
3 0.773643 0.573366 0.573366 0.838784 1.000000
bindendes
p-MO
11
2.000000
1 0.000000
2 1.000000
3 1.000000
----- POPULATIONS IN EACH AO -----
MULLIKEN LOWDIN
1 C 1 S 1.99735 1.99085
2 C 1 S 0.99476 1.01616
3 C 1 X 0.85327 0.86023
4 C 1 Y 0.83590 0.93469
5 C 1 Z 0.85327 0.86023
6 O 2 S 1.99880 1.99803
7 O 2 S 1.86626 1.74370
8 O 2 X 1.57337 1.56989
9 O 2 Y 1.22094 1.28742
10 O 2 Z 1.57337 1.56989
11 O 3 S 1.99880 1.99803
12 O 3 S 1.86626 1.74370
13 O 3 X 1.57337 1.56989
14 O 3 Y 1.22094 1.28742
15 O 3 Z 1.57337 1.56989
----- MULLIKEN ATOMIC OVERLAP POPULATIONS -----
(OFF-DIAGONAL ELEMENTS NEED TO BE MULTIPLIED BY 2)
1
2 3
1 4.6383405
2 0.4481060 7.7915860
3 0.4481060
-0.0069681 7.7915860
TOTAL MULLIKEN AND LOWDIN ATOMIC POPULATIONS
ATOM MULL.POP. CHARGE LOW.POP. CHARGE
1 C 5.534552 0.465448 5.662150 0.337850
2 O 8.232724 -0.232724 8.168925 -0.168925
3 O 8.232724 -0.232724 8.168925 -0.168925
-------------------------------
BOND ORDER AND VALENCE ANALYSIS BOND ORDER THRESHOLD=0.050
-------------------------------
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM
PAIR DIST ORDER
1 2 1.187 1.965
1 3 1.187 1.965 2 3
2.373 0.385
zum Vergleich SiO2:
BOND
BOND
BOND
ATOM PAIR DIST ORDER ATOM PAIR DIST ORDER ATOM PAIR
DIST ORDER
1 2 1.573
1.840 1 3
1.573 1.840 2 3
3.147 0.349
TOTAL BONDED FREE
ATOM VALENCE VALENCE VALENCE
1 C 3.930 3.930 0.000
2 O 2.350 2.350 0.000
3 O 2.350 2.350 0.000
---------------------
ELECTROSTATIC MOMENTS
---------------------
POINT 1 X Y Z (BOHR) CHARGE
0.000000 0.000000 0.000000 0.00 (A.U.)
DX DY DZ /D/ (DEBYE)
0.000000 0.000000 0.000000 0.000000
...... END OF PROPERTY EVALUATION ......